









砂粒と星々は永久に回転し続ける一瞬の相似形である。
我々は地上を這いて、天の物語を紡ぐ。
誰にでもなれるが、誰でもは選べない。
□ Matlis category equivalences for a ring epimorphism
>> https://arxiv.org/pdf/1907.04973v1.pdf
The triangulated Matlis equivalence is an equivalence between the (bounded or unbounded) derived category of complexes R-modules with u-comodule cohomology modules and the similar derived category of complexes of R-modules w/ u-contramodule cohomology modules.
Further assumptions allow to describe the third category in the recollement as the unbounded derived category of the abelian categories of u-comodules & u-contramodules.
For commutative rings, any homological epimorphism of projective dimension 1 is flat. Injectivity of the map u is not required.

□ Conos: Joint analysis of heterogeneous single-cell RNA-seq dataset collections
>> https://www.nature.com/articles/s41592-019-0466-z
Conos, an approach that relies on multiple plausible inter-sample mappings to construct a global graph connecting all measured cells.
The graph enables identification of recurrent cell clusters and propagation of information between datasets in multi-sample or atlas-scale collections.
□ Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression
>> https://www.nature.com/articles/s41588-019-0462-3
These extensive rearrangements caused many changes to chromatin topology, disrupting long-range loops, topologically associating domains (TADs) and promoter interactions, yet these are not predictive of changes in expression.
Gene expression is generally not altered around inversion breakpoints, indicating that mis-appropriate enhancer–promoter activation is a rare event.
Similarly, shuffling or fusing TADs, changing intra-TAD connections and disrupting long-range inter-TAD loops does not alter expression for the majority of genes.
□ VariantQC: a visual quality control report for variant evaluation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz560/5532508
To ensure variant and genotype data are consistent and accurate, it is necessary to evaluate variants prior to downstream analysis using quality control (QC) reports.
DISCVR-seq Toolkit is a diverse collection of tools for working with sequencing data, developed and maintained by the Bimber Lab, built using the GATK4 engine.

□ Dirac mixture distributions for the approximation of mixed effects models
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/16/703850.full.pdf
a first scalability analysis for Cram ́er-von Mises Distance (CMD) methods as well as a comparison with MC, QMC and SP methods for the analysis of nonlinear MEMs.
In contrast to sigma-point methods, the method based on the modified Cram ́er-von Mises Distance allows for a flexible number of points and a more accurate approximation for nonlinear problems.

□ Visualization and analysis of RNA-Seq assembly graphs
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz599/5532290
The resulting graphs are visualized in 3D space to better appreciate their sometimes large and complex topology, with other information being overlaid on to nodes, e.g. transcript models.
Demonsrating the utility of this approach, including the unusual structure of these graphs and how they can be used to identify issues in assembly, repetitive sequences within transcripts and splice variants.
the data pipeline, the tools and basic approach presented here provide an effective analytical paradigm that is a novel contribution to the analysis of the huge amounts of information-rich but complex data produced by modern DNA sequencing platforms.
□ Integrative prediction of gene expression with chromatin accessibility and conformation data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/16/704478.full.pdf
an extension of the Tepic framework to account for PEIs inferred from chromatin conformation capture experiments.
This novel machine learning approach that allows to prioritize TFs in distal loop and promoter regions with respect to their importance for GE regulation.
□ CNAPE: A Machine Learning Method for Copy Number Alteration Prediction from Gene Expression
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/17/704486.full.pdf
CNAPE takes a prior knowledge-aided multinomial logistic regression wi/ LASSO to predict CNA, which differs from canonical DNA-seq based methods in that CNAPE first selects/inputs genes whose expression levels are responsive to copy number change, and then build regression models on these genes.
The results from CNAPE would also be valuable to recalibrate tools such as GISTIC to identify regions with significant copy number aberrations from large cohorts.
□ Janus: An Extensible Open-Source Software Package for Adaptive QM/MM Methods
>> https://pubs.acs.org/doi/10.1021/acs.jctc.9b00182
Adaptive quantum mechanics/molecular mechanics (QM/MM) approaches are able to treat systems with dynamic or nonlocalized active centers by allowing for on-the-fly reassignment of the QM region.
Janus currently interfaces with Psi4 and OpenMM, but its modular infrastructure enables easy extensibility to other molecular codes without major modifications to either code.

□ Long live the king: chromosome-level assembly of the lion (Panthera leo) using linked-read, Hi-C, and long read data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/17/705483.full.pdf
This assembly is composed of 10x Genomics Chromium data, Dovetail Hi-C, and Oxford Nanopore long-read data.
The quality of this assembly allowed us to investigate the co-linearity of the genome compared to other felids and the importance of the reference sequence for estimating heterozygosity.

□ MPLNClust: A multivariate Poisson-log normal mixture model for clustering transcriptome sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2916-0
Parameter estimation is carried out using a Markov chain Monte Carlo expectation-maximization (MCMC-EM) algorithm, and information criteria are used for model selection.
the hidden layer of the MPLN distribution is a multivariate Gaussian distribution, which allows for the specification of a covariance structure.
□ RevMet: Semi‐quantitative characterisation of mixed pollen samples using MinION sequencing and Reverse Metagenomics
>> https://besjournals.onlinelibrary.wiley.com/doi/10.1111/2041-210X.13265
RevMet (Reverse Metagenomics), that allows reliable and semi‐quantitative characterization of the species composition of mixed‐species eukaryote samples, such as bee‐collected pollen, without requiring reference genomes.
RevMet can identify plant species present in mixed‐species samples at proportions of DNA ≥1%, with few false positives and false negatives, and reliably differentiate species represented by high versus low amounts of DNA in a sample.
□ DeepExpression: Integrating distal and proximal information to predict gene expression via a densely connected convolutional neural network
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz562/5535598
DeepExpression, a densely connected convolutional neural network to predict gene expression using both promoter sequences and enhancer-promoter interactions.
DeepExpressiom consistently outperforms baseline methods not only in the classification of binary gene expression status but also in the regression of continuous gene expression levels, in both cross-validation experiments & cross-cell lines predictions.

□ TREEasy: an automated workflow to infer gene trees, species trees, and phylogenetic networks from multilocus data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/18/706390.full.pdf
TREEasy, that performs automated sequence alignment (with MAFFT), gene tree inference, species inference from concatenated data (with IQ-Tree), species tree inference from gene trees (with ASTRAL, MP-EST, and STELLS2), and phylogenetic network inference (with SNaQ and PhyloNet).
un-rooted selected gene trees generated by IQ-TREE are put together as input to infer a species tree using ASTRAL.
Meanwhile, the un-rooted gene trees are rooted with a preset parameter R (species name(s)) and then the rooted gene trees are used to infer species trees using STELLS2 and MP-EST.

□ OMA standalone: orthology inference among public and custom genomes and transcriptomes
>> https://genome.cshlp.org/content/29/7/1152.abstract
The Orthologous MAtrix (OMA) database is a leading resource for identifying orthologs among publicly available, complete genomes. Here, we describe the OMA pipeline available as a standalone program.
When run on a cluster, it has native support for the LSF, SGE, PBS Pro, and Slurm job schedulers and can scale up to thousands of parallel processes.
Another key feature of OMA standalone is that users can combine their own data with existing public data by exporting genomes and precomputed alignments from the OMA database, which currently contains over 2100 complete genomes.

□ Machine and deep learning meet genome-scale metabolic modeling
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007084
A multiview approach merging experimental and knowledge-driven omic data through machine learning methods can incorporate key mechanistic information in an otherwise biologically-agnostic learning process.
Mining and integrating experimental and GSMM-generated multiomic data with machine learning techniques can unveil unknown mechanisms in a sample-specific manner, hence identifying relevant targets for biotechnology and biomedicine.
□ Docker4Circ: A Framework for a Reproducible Characterization of CircRNAs from RNA-Seq Data
>> https://www.preprints.org/manuscript/201907.0219/v1
CircRNAs are widely expressed in both cancerous and normal tissues [30,31] and an increased number of sequencing experiments is becoming accessible to explore circRNAs expression in a specific biological context.
Docker4Circ a comprehensive framework for circRNAs analysis merging four different modules into a reproducible analysis framework from circRNAs prediction to their expression analysis.
□ TGStools: A Bioinformatics Suit to Facilitate Transcriptome Analysis of Long Reads from Third Generation Sequencing Platform
>> https://www.mdpi.com/2073-4425/10/7/519
TGStools, a package that implements multiple tools to facilitate routine transcriptome analysis, such as isoforms comparison, detecting alternative splicing (AS) pattern and lncRNAs identification.
In the ‘Transcripts’ category, the tool ‘TransDisp’ compares the isoforms of the queried gene and displays the sequenced transcripts along with multiple genomic annotations; ‘StaDist’ automatically finds the nearby genomics feature and calculates the distance;
in the ‘Alternative splicing’ category, ‘StaAS’ identifies the alternative events and detects the difference of each alternative splicing event among samples; ‘CalScoreD’ selects the most spliced genes; ‘GOEnrich’ selects top ranked gene ontology terms which are enriched with the most spliced genes.

□ C-InterSecture—a computational tool for interspecies comparison of genome architecture
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz415/5497251
C-InterSecture (Computional tool for InterSpecies analysis of genome architecture) pipeline is python 2.7 based utilits to cross-species comparison of Hi-C map.
C-InterSecture was designed to liftover contacts between species, compare 3-dimensional organization of defined genomic regions, such as TADs, and analyze statistically individual contact frequencies.
C-InterSecture allows statistical comparison of contact frequencies of individual pairs of loci, as well as interspecies comparison of contacts pattern within defined genomic regions, i.e. topologically associated domains.

□ CWL-Airflow: a lightweight pipeline manager supporting Common Workflow Language
>> https://academic.oup.com/gigascience/article/8/7/giz084/5535758
CWL-Airflow uses CWL version 1.0 specification and can run workflows on stand-alone MacOS/Linux servers, on clusters, or on a variety of cloud platforms.
CWL-Airflow uses a CWL version of a Python pipeline from BioWardrobe.
□ Robustifying genomic classifiers to batch effects via ensemble learning
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/20/703587.full.pdf
The first developed prediction models within each batch, then integrate them through ensemble weighting methods.
observing a turning point in the level of heterogeneity, after which this strategy of integrating predictions yields better discrimination in independent validation than the traditional method of integrating the data.

□ The MultiOmics Explainer: explaining omics results in the context of a pathway/genome database
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2971-6
the MultiOmics Explainer searches the organism’s network of metabolic reactions, transporters, cofactors, enzyme substrate-level activation and inhibition relationships, and transcriptional and translational regulation relationships to identify paths of influence among input genes.
This approaches to graph construction are quite different however—their approach is more computationally demanding but results in a complete set of possible paths (which then must be prioritized), whereas using cutoffs and other heuristics to reduce computation time and limit the number of paths produced.
□ A massively parallel algorithm for finding non-existing sequences in genomes
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/20/709949.full.pdf
an algorithm for a given reference genome, a set of sufficiently long absent words in that genome (>= 18) with a guaranteed Hamming distance along all positions of the reference and additional information about the number of mismatches.
Meta-heuristics and parallel implementations with good practical running times have also been developed; the drawback of these approaches is that they cannot guarantee that an exact solution will be found.
□ pyBedGraph: a Python package for fast operations on 1-dimensional genomic signal tracks
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/20/709683.full.pdf
As genomics researchers continue to develop novel technologies ranging from bulk cells to single-cell and single-molecule experiments, it will be imperative to distinguish true signal from technical noise.
When tested on 8 ChIP-seq and ATAC-seq datasets, pyBedGraph is on average 245 times faster than the existing program. Notably, pyBedGraph can look up the exact mean signal of 1 million regions in ~0.26 second on a conventional laptop.

□ REVERSE ENGINEERING GENE NETWORKS USING GLOBAL-LOCAL SHRINKAGE RULES
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/21/709741.full.pdf
The proposed method handles heavy-tailed data by assuming a multivariate heavy-tailed data likelihood that mixes over Gaussian variance components.
the proposed method performs well in the high-dimensional situation, and justify its use in this case by providing sufficient conditions for posterior propriety.

□ DeepResolve: Visualizing complex feature interactions and feature sharing in genomic deep neural networks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2957-4
DeepResolve is capable of visualizing complex feature contribution patterns and feature interactions that contribute to decision making in genomic deep convolutional networks.
DeepResolve reveals that DeepSEA’s learned decision structure is shared across genome annotations including histone marks, DNase hypersensitivity, and transcription factor binding.

□ Wx: a neural network-based feature selection algorithm for transcriptomic data
>> https://www.nature.com/articles/s41598-019-47016-8
The Wx algorithm ranks genes based on the discriminative index (DI) score that represents the classification power for distinguishing given groups.
The proposed feature selection method was based on softmax regression, which utilizes a simple one-layer neural network regression model in which the dependent variable is categorical.
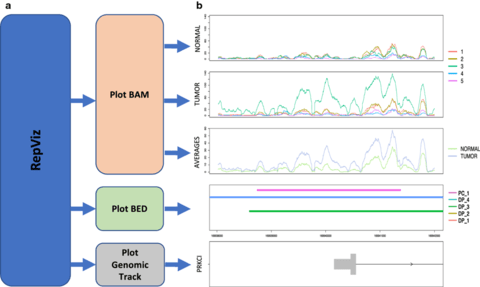
□ RepViz: a replicate-driven R tool for visualizing genomic regions
>> https://bmcresnotes.biomedcentral.com/articles/10.1186/s13104-019-4473-z
RepViz - replicate-driven visualization allows simultaneous viewing of both intra- and intergroup variation in sequencing counts of the studied conditions, as well as their comparison to the output features (e.g. identified peaks) from user selected analysis methods.
The RepViz tool is primarily designed for chromatin data, such as ChIP-seq and ATAC-seq, but can also be used with other sequencing data, such as RNA-seq, or combinations of different types of genomic data.
□ ChromSCape : an R/Shiny application for interactive analysis of single-cell chromatin profiles
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/22/683037.full.pdf
The pipeline is designed for high-throughput single-cell datasets with samples containing as low as 100 cells and with a minimum of 1000 reads per cell.
The interactive process includes filtering out cells with low coverage and regions, dimensionality reduction by PCA, classifying cells in an unsupervised manner to identify sub-populations and find biologically relevant loci differentially enriched in each sub-populations.

□ SquiggleKit: A toolkit for manipulating nanopore signal data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz586/5537108
SquiggleKit can be used to facilitate data management, to generate fine-tuned datasets for machine learning, to visualise signal, to validate demultiplexing results, and to identify motifs of interest without base calling, amongst other applications.
Targeting regions of interest in raw signal data: Segmenter identifies the boundaries of relatively long regions of signal attenuation.
MotifSeq takes a query nucleotide sequence as input, converts it to a normalised signal trace (i.e., ’events’), then performs signal-level local alignment using a dynamic programming algorithm.
□ GSMA: an approach to identify robust global and test Gene Signatures using Meta-Analysis
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz561/5536878
GSMA, an intra- and inter-level meta-analysis framework that overcomes these limitations and provides a gene signature that is reliable and reproducible across multiple independent studies of a given disease.
The approach provides a comprehensive global signature that can be used to understand the underlying biological phenomena, and a smaller test signature that can be used to classify future samples of a given disease.

□ A hierarchical Bayesian mixture model for distinguishing active gene expression from transcriptional noise
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/23/711630.full.pdf
a Bayesian approach to infer the parameters of the hierarchical mixture model from patterns of relative expression across a set of replicate RNA-seq libraries, providing estimates of the posterior probability that each gene in the genome is actively expressed in a given tissue or cell type.
Posterior-predictive simulation suggests that this model fits diverse datasets and provide a means of measuring model fit for future improvements of this method.
□ PgRC: Pseudogenome based Read Compressor
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/23/710822.full.pdf
Pseudogenome-based Read Compressor (PgRC), an in-memory algorithm for compressing the DNA stream, based on the idea of building an approximation of the shortest common superstring over high-quality reads.
PgRC wins in compression ratio over its main competitors, SPRING and Minicom, by up to 18 and 21 percent on average, respectively, while being at least comparably fast in decompression.
A crucial, but also often most time-consuming phase of PgRC compression is building the pseudogenomes. Orthogonal to a parallel architecture, a major boost is perhaps possible here due to algorithmic improvements.

□ RaPID: ultra-fast, powerful, and accurate detection of segments identical by descent (IBD) in biobank-scale cohorts
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1754-8
The key idea of RaPID is that the problem of approxi- mate high-resolution matching over a long range can be mapped to the problem of exact matching of low- resolution subsampled sequences with high probability.
RaPID achieves a time and space complexity linear to the input size and the number of reported IBDs.
□ FLASHDeconv: Ultrafast, high-quality feature deconvolution for top-down proteomics
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/25/714915.full.pdf
FLASHDeconv, an algorithm based on a simple transformation of mass spectra, which turns deconvolution into the search for constant patterns thus greatly accelerating the process.
The major speed-up of FLASHDeconv is achieved by very fast decharging (i.e., assigning charges to peaks) in the spectral deconvolution step.
□ Accuracy of de novo assembly of DNA sequences from double-digest libraries varies substantially among software
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/18/706531.full.pdf
ABySS failed to recover any true genome fragments, and Velvet and VSEARCH performed poorly for most simulations. Stacks, Stacks2, and CD-HIT recovered a high proportion of true fragments and produced accurate assemblies of simulations containing SNPs.
comparing the completeness of the assemblies (fraction of all true genome fragments represented) and their degree of over-assembly (i.e., collapsing multicopy, paralogous loci into a single contig) and under-assembly (i.e., separating allelic variants at a single locus into different contigs).

□ DNBseq: Impact of sequencing depth and technology on de novo RNA-Seq assembly
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5965-x
The missing “gap” regions in the HiSeq assemblies were often attributed to higher GC contents, but this may be an artefact of library preparation and not of sequencing technology.
Increasing sequencing depth beyond modest data sets of less than 10 Gbp recovers a plethora of single-exon transcripts undocumented in genome annotations.

□ Efficient parameterization of large-scale dynamic models based on relative measurements
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz581/5538985
a novel hierarchical approach combining the efficient analytic evaluation of optimal scaling, offset, and error model parameters with the scalable evaluation of objective function gradients using adjoint sensitivity analysis.
This hierarchical formulation is applicable to a wide range of models, and allows for the efficient parameterization of large-scale models based on heterogeneous relative measurements.
□ DDE_BD: Bayesian inference of distributed time delay in transcriptional and translational regulation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz574/5538987
a statistical inference method in order to estimate reaction constants of simple Birth-Death process with time delay.
Although the resulting models are non-Markovian, recent results on stochastic systems with random delays allow us to rigorously obtain expressions for the likelihoods of model parameters.
this allows us to extend MCMC methods to efficiently estimate reaction rates, and delay distribution parameters, from single-cell assays.










※コメント投稿者のブログIDはブログ作成者のみに通知されます