(Created with Midjourney v7)
□ The Ambientalist / “Spica”




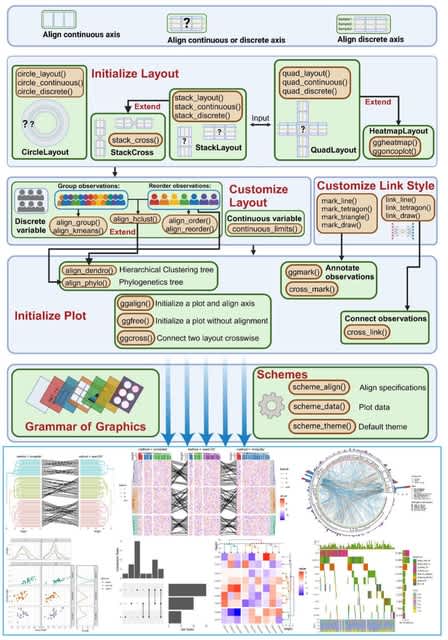
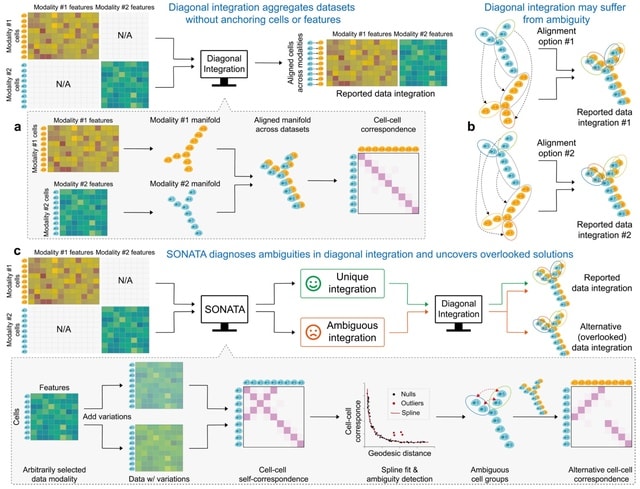
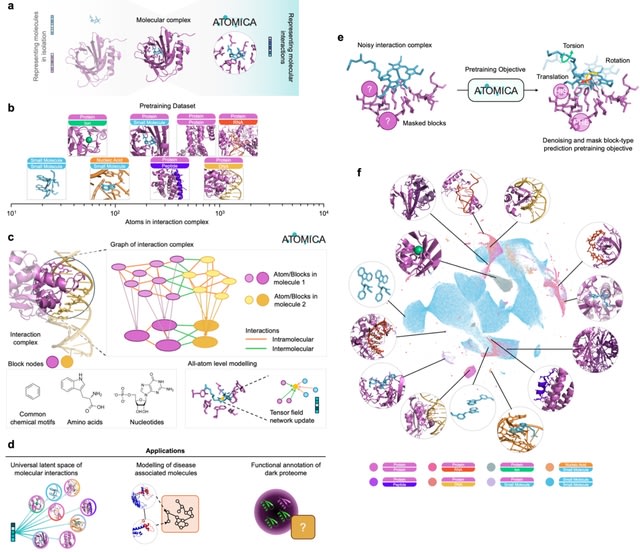
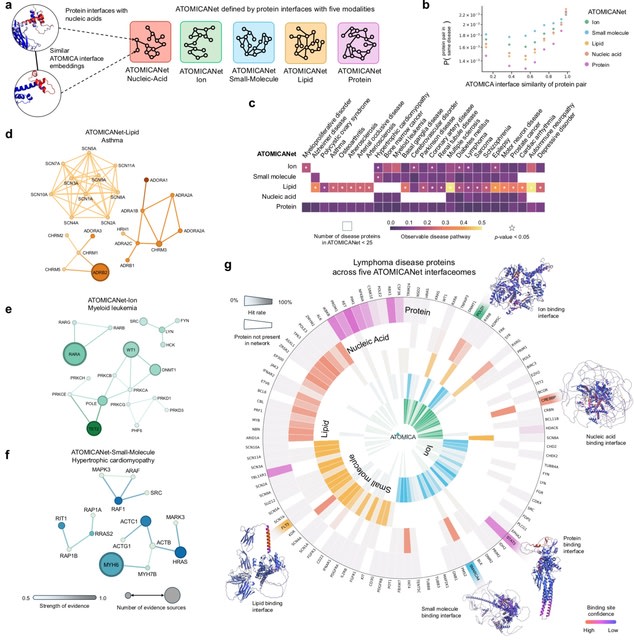
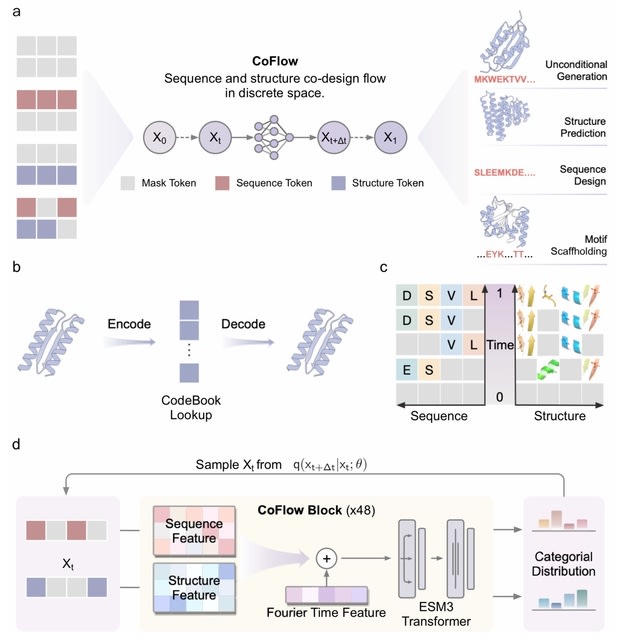
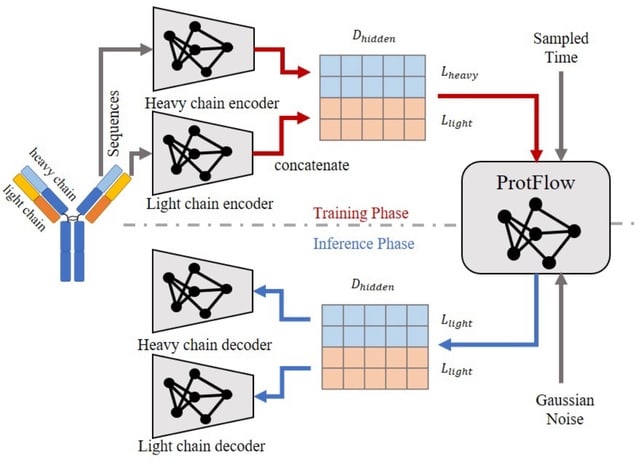
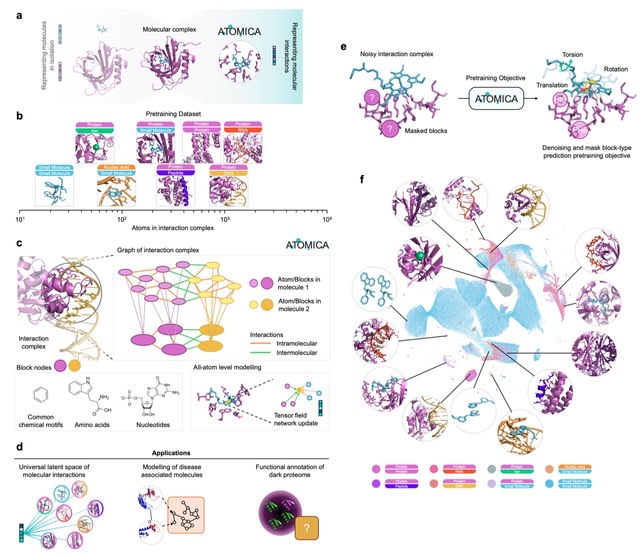
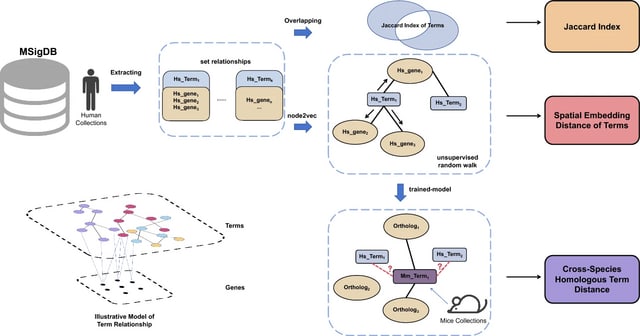
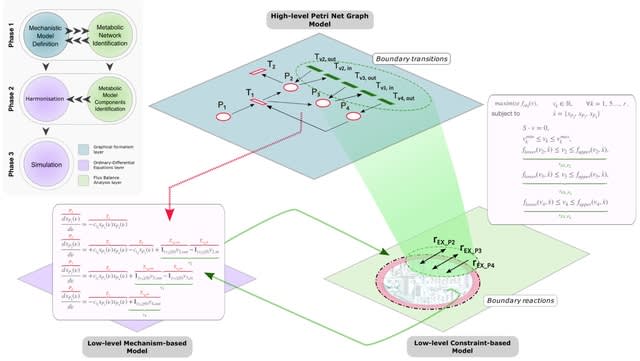
□ Cosmos: A Position-Resolution Causal Model for Direct and Indirect Effects in Protein Functions
>> https://www.biorxiv.org/content/10.1101/2025.08.01.667517v1
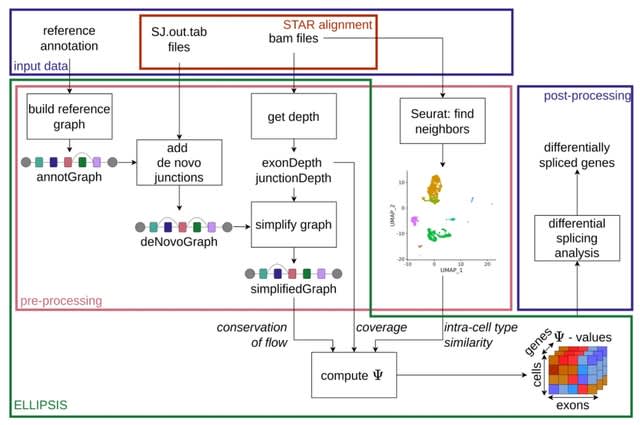
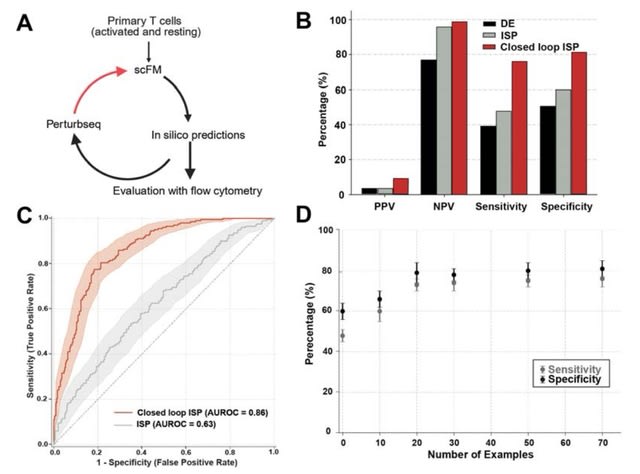
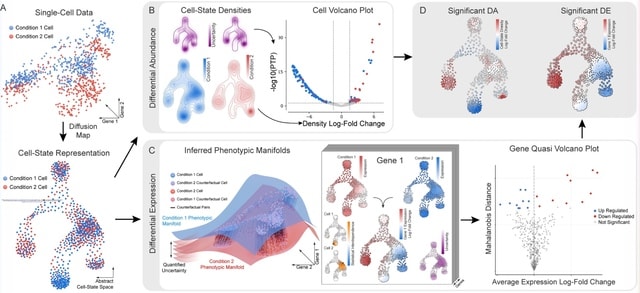
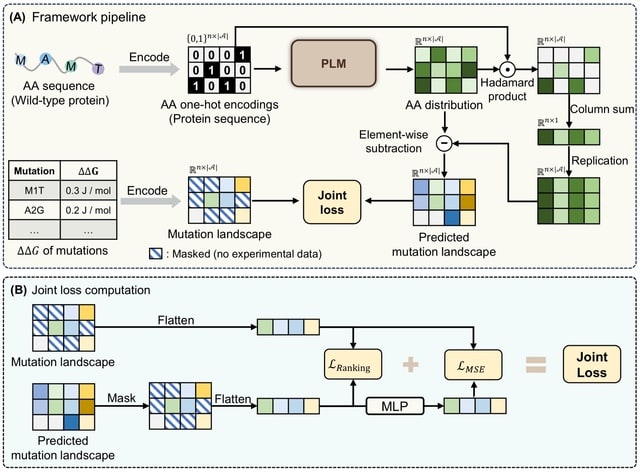
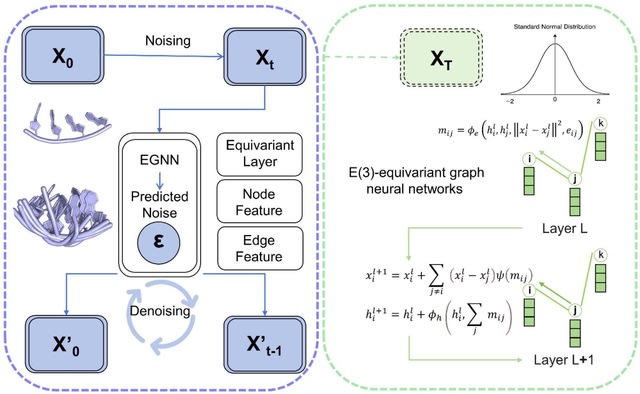
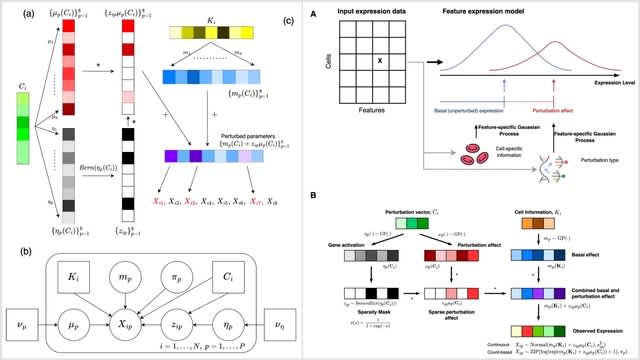
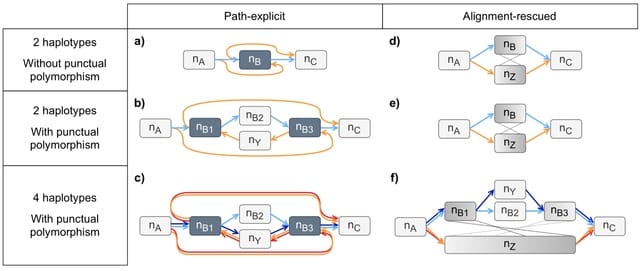
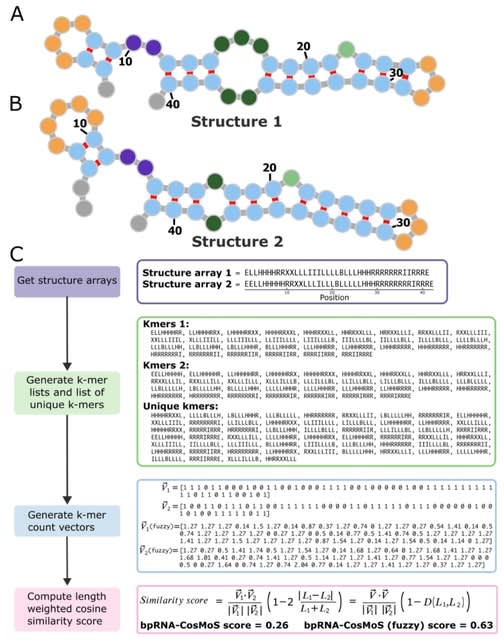
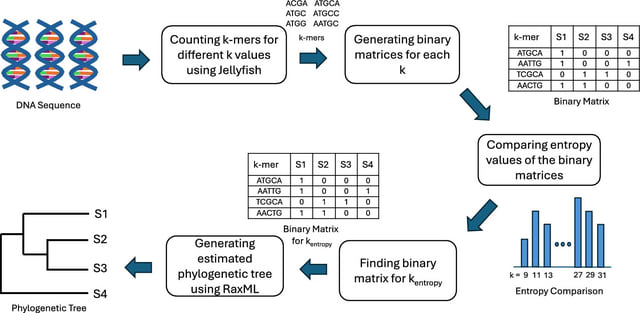
Cosmos, a Bayesian model selection framework designed to support causal inference between related phenotypes in Deep Mutational Scanning data with single mutations. It determines whether a relationship exists between two phenotypes and estimates the strength of that relationship.
Cosmos generates counterfactual predictions of what would happen to the downstream phenotype if the upstream phenotype were fixed to a reference value. Cosmos uses position-level aggregation and Bayesian model selection to infer interpretable causal structures.




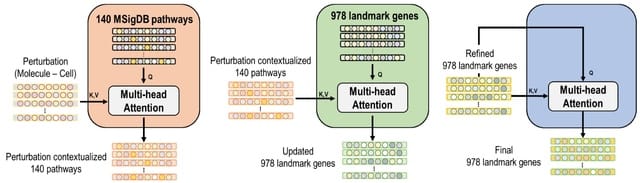
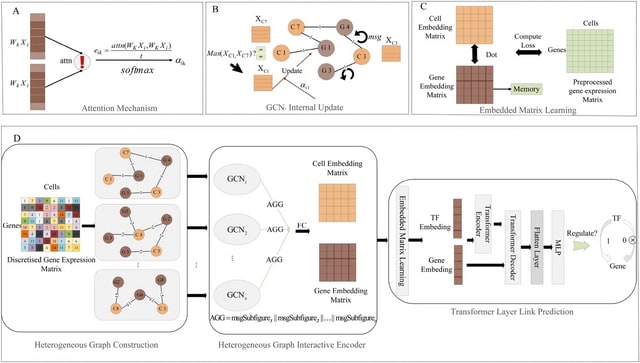
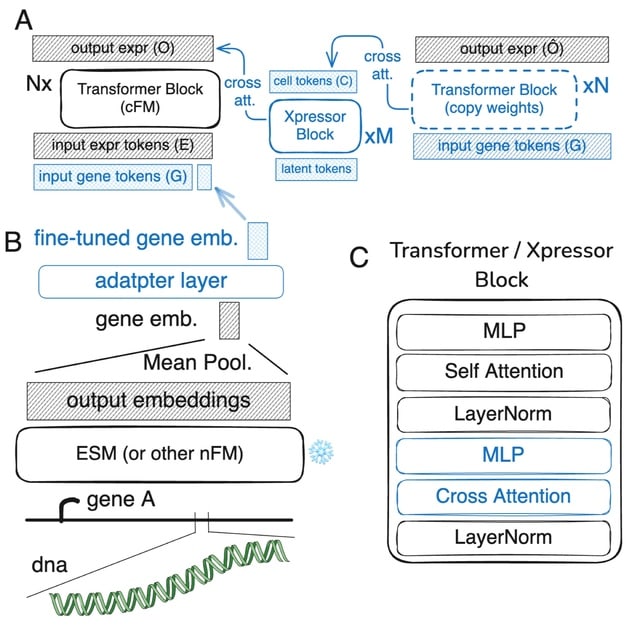
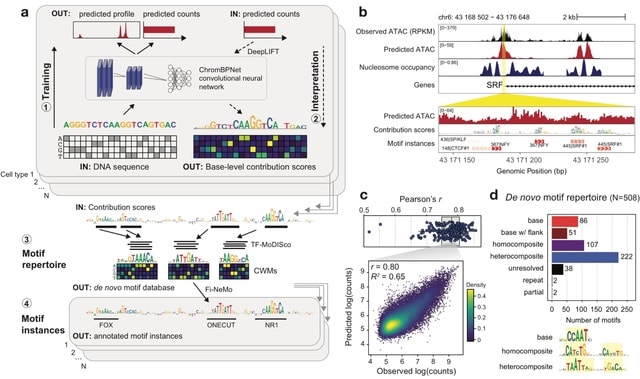
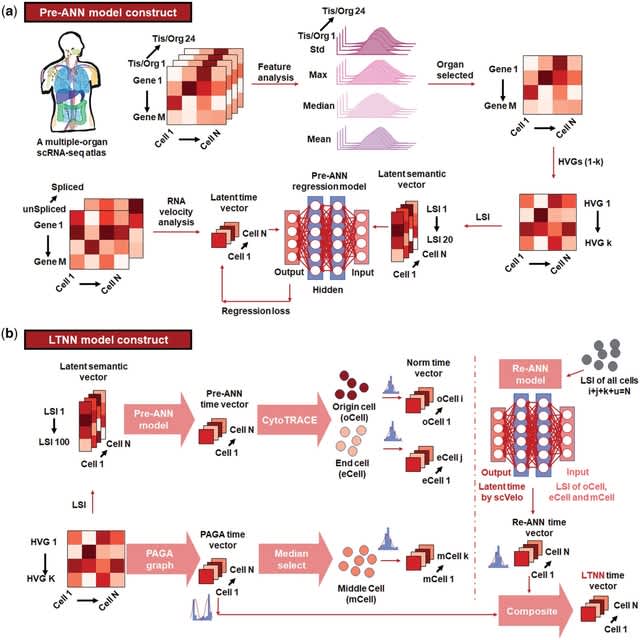
□ CellForge: Agentic Design of Virtual Cell Models
>> https://arxiv.org/abs/2508.02276
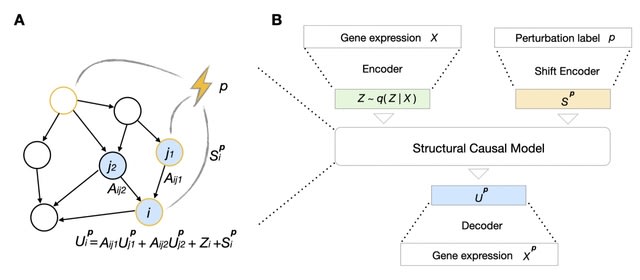
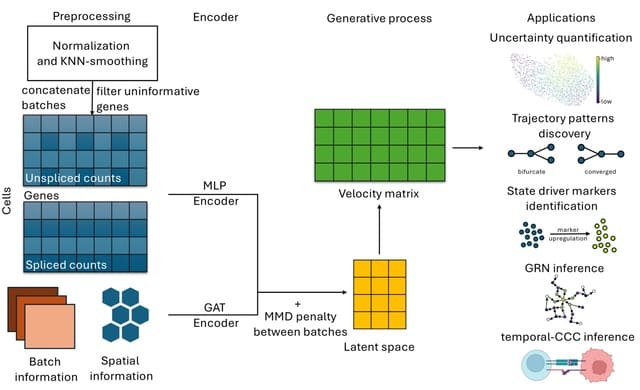
CELLFORGE, an agentic system that leverages a multi-agent framework that transforms presented biological datasets and research objectives directly into optimized computational models for virtual cells. CELLFORGE outputs both an optimized model architecture and executable code.
CELLFORGE confronts the interdisciplinary complexity of virtual-cell modelling by casting the entire research cycle as a collaboration between role-specialised agents. TaskAnalysis agents begin by profiling the dataset and mining the literature, distilling a draft research plan.
Design agents engage in a graph-structured debate, iteratively proposing, critiquing, and fusing candidate architectures until the cohort converges on an optimised model and experimental protocol. Experiment-Execution agents translate this plan into runnable code.

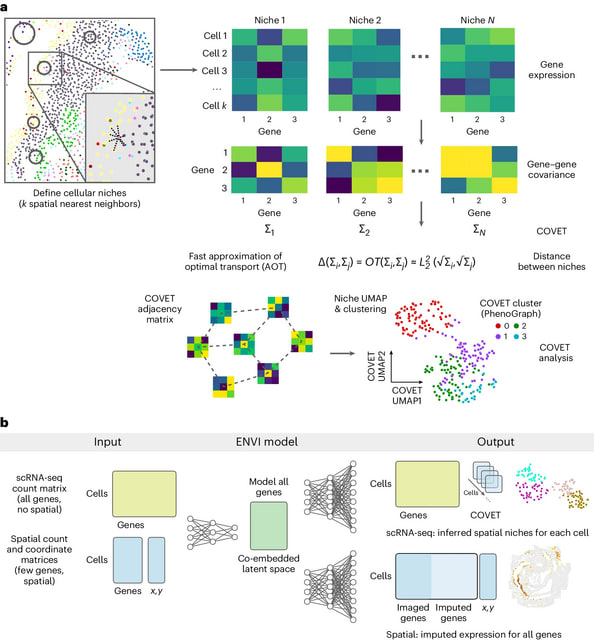
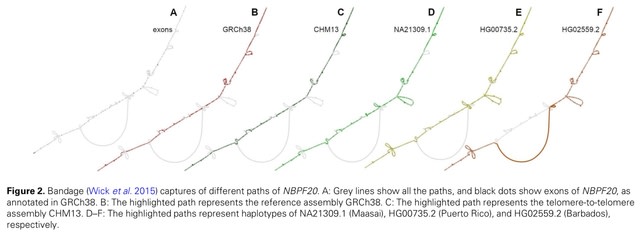
□ DNARetrace: DNA Sequence Trace Reconstruction Using Deep Learning
>> https://www.biorxiv.org/content/10.1101/2025.08.05.668822v1
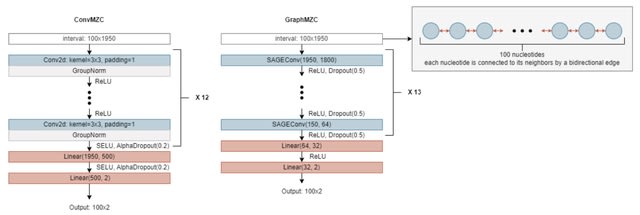
DNARetrace is a DNA sequence trace reconstruction model that performs preprocessing and dataset construction, and then employs a Bidirectional Fourier-Kolmogorov-Arnold Network (Bi-FKGAT), using an extremely unbalanced loss function for link prediction.
DNARetrace addresses the unidirectional neighborhood aggregation defect of GNN studies. It achieves the automatic conversion of data into graph structure by integrating multi-platform sequence alignment tools, diverse DNA fragment graph generation, and labeling of DNA fragment.

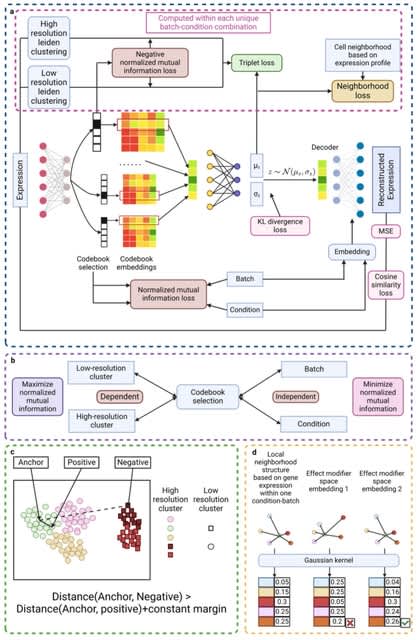
□ BioScientist Agent: Designing LLM-Biomedical Agents with KG-Augmented RL Reasoning Modules for Drug Repurposing and Mechanistic of Action Elucidation
>> https://www.biorxiv.org/content/10.1101/2025.08.08.669291v1
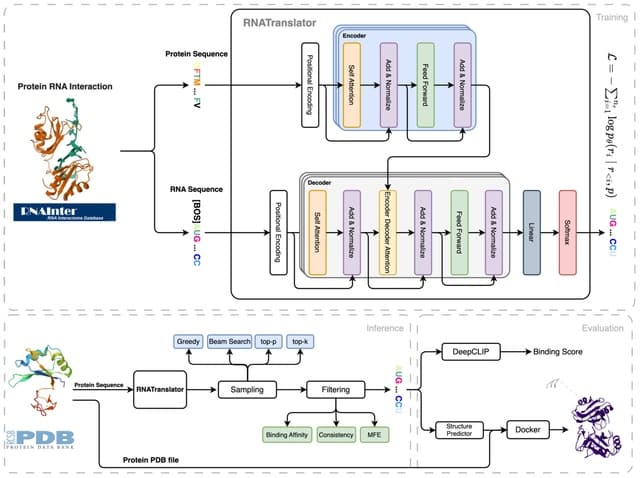
BioScientist Agent, an end to end framework that unifies a billion-fact biomedical knowledge graph with a variational graph auto-encoder for representation learning and link prediction driven repositioning.
BioScientist Agent uses a reinforcement learning module that traverses the graph to recover biologically plausible mechanistic paths. A LLM multi-agent layer enables inference of target pathways for a drug disease pair, and automatic generation of coherent causal reports.

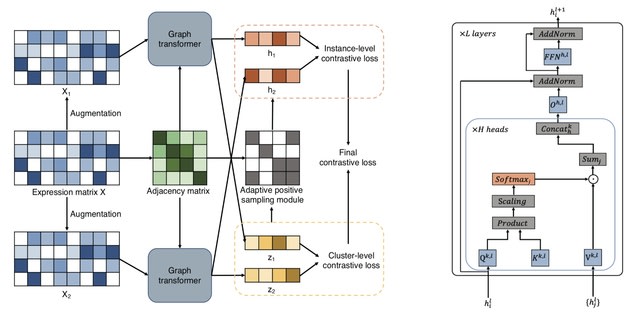
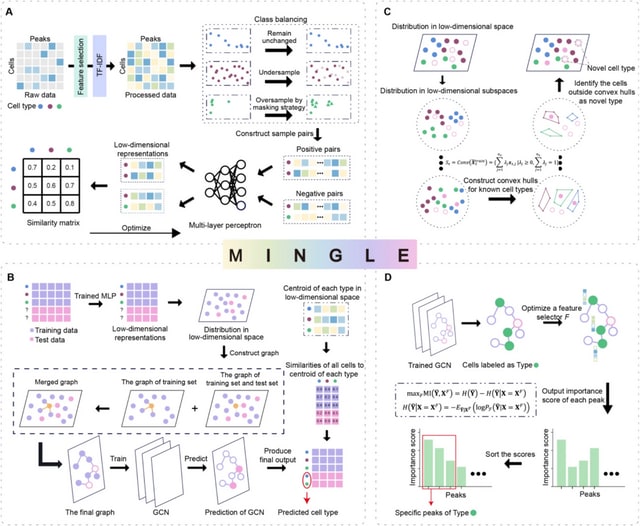
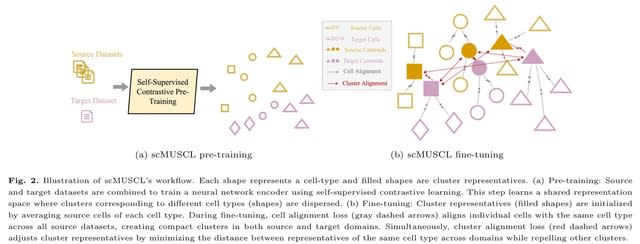
□ Less is more: Improving cell-type identification with augmentation-free single-cell RNA-Seq contrastive learning (AF-RCL)
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf437/8222716
AF-RCL creates one pair of positive and negative cell sets. The positive cell set consists of those cells belonging to the same cell-type as the target cell, whilst all other cells belonging to different cell-types to the target cell are included in the negative cell set.
Those different pairs of positive and negative cell sets are then used as inputs for two neural networks (i.e. an encoder and a projector) to learn the discriminative feature representations using a modified contrastive learning loss function, without any data augmentation operation.

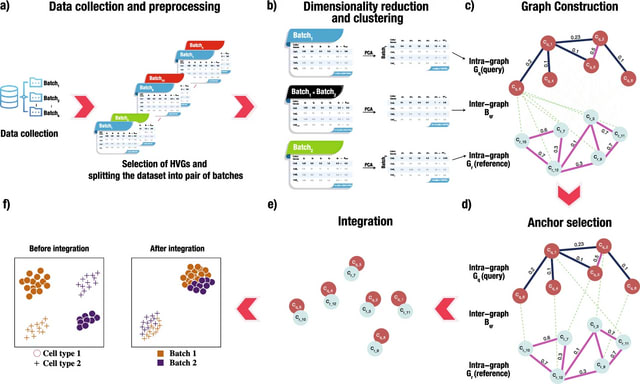
□ scECDA: Multi-omics single-cell data alignment and integration with enhanced contrastive learning and differential attention mechanism
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf443/8224605
scECDA, a novel approach for single-cell multi-omics data alignment and integration. scECDA incorporates a differential attention mechanism and introduces a feature fusion module that automatically enhances the signal-to-noise ratio of biologically relevant features.
scECDA employs contrastive learning alongside a simple yet effective data augmentation strategy to generate positive and negative samples. scECDA directly outputs both the integrated latent representation of multi-omics data and the final cell clustering assignments.

□ Hi-Cformer enables multi-scale chromatin contact map modeling for single-cell Hi-C data analysis
>> https://www.biorxiv.org/content/10.1101/2025.08.04.668453v1
Hi-Cformer, a transformer-based method that simultaneously models multi-scale blocks of chromatin contact maps and incorporates a specially designed attention mechanism to capture the dependencies between chromatin interactions across genomic regions and scales.
Hi-Cformer robustly derives low-dimensional representations of cells from single-cell Hi-C data, achieving clearer separation of cell types. Hi-Cformer imputes chromatin interaction signals associated with cellular heterogeneity, incl. TAD-like boundaries and A/B compartments.

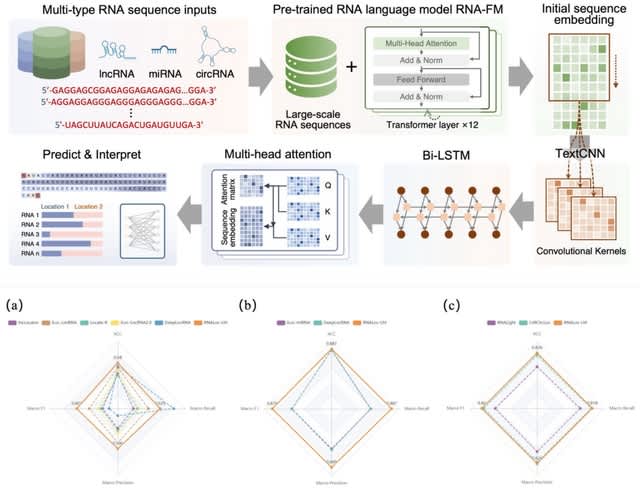
□ structRFM: A fully-open structure-guided RNA foundation model for robust structural and functional inference
>> https://www.biorxiv.org/content/10.1101/2025.08.06.668731v1
structRFM, a structure-guided RNA foundation model that is pre-trained on millions of RNA sequences and secondary structures data by integrating base pairing interactions into masked language modeling through a novel pair matching operation.
structRFM employs an elaborately designed structure-guided masked language modeling (SgMLM) strategy. SgMLM is a structure-guided pre-training strategy, featuring two core components: structure-guided masking and dynamic masking balance.
structRFM selectively masks input tokens corresponding to canonical base pairs within local structural contexts, encouraging the model to recover base-pair interactions based on neighboring loop regions. structRFM balances nucleotide-wise and structure-wise masking.

□ Longdust: Identify long STRs, VNTRs, satellite DNA and other low-complexity regions in a genome
>> https://github.com/lh3/longdust
Longdust identifies long highly repetitive STRs, VNTRs, satellite DNA and other low-complexity regions (LCRs) in a genome. It is motivated by and follows a similar rationale to SDUST. Longdust can find centromeric satellite and VNTRs with long repeat units.
Longdust overlaps with tandem repeat finders (e.g. TRF, TANTAN and ULTRA) in functionality. Nonetheless, it is not tuned for tandem repeats with two or three copies, but may report low-complexity regions without clear tandem structure. Longdust complements TRF etc to some extent.
Longdust uses BLAST-like X-drop to break at long non-LCR intervals. Due to heuristics, Longdust generates slightly different output on the reverse complement of the input sequence. For strand symmetry like SDUST, Longdust takes the union of intervals identified from both strands.

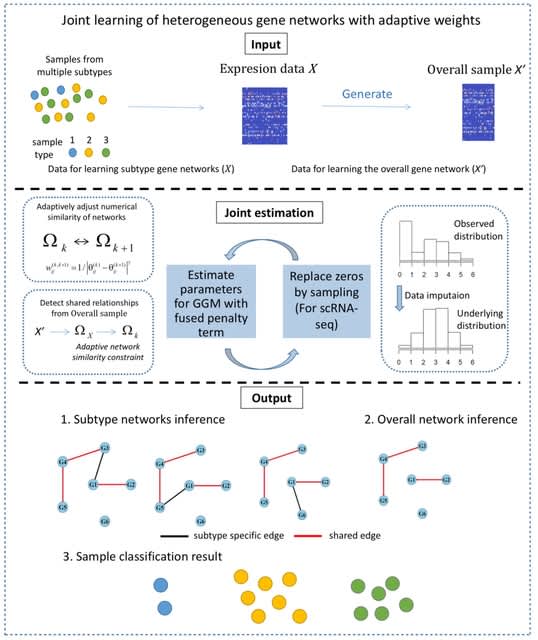
□ MOH: a novel multilayer multi-omics heterogeneous graph for single-cell clustering
>> https://www.biorxiv.org/content/10.1101/2025.08.04.668248v1
MOH constructs a multilayer heterogeneous graph to simultaneously extract and enhance representations from all three omics layers, incorporating both intra-layer and inter-layer edges to capture association and similarity relationships.
MOH use Deep Graph Infomax (DGI), an unsupervised graph embedding method, to learn node representations from graph-structured data. It maximizes the mutual information b/n global and local representations of the graph. The features extracted by DGI include both local and global.

□ TPClust: Temporal Profile-Guided Subtyping Using High-Dimensional Omics Data
>> https://www.biorxiv.org/content/10.1101/2025.08.05.668514v1
TPClust, a supervised, semi-parametric clustering method that integrates high-dimensional omics data with longitudinal phenotypes including outcomes and covariates for outcome-guided subtyping.
TPClust models latent subtype membership / longitudinal outcome trajectories using multinomial logistic regression informed by molecular features selected via structured regularization, along w/ spline-based regression to capture subtype-specific, time-varying covariate effects.


□ scTail: precise polyadenylation site detection and its alternative usage analysis from reads 1 preserved 3′ scRNA-seq data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03710-7
scTail identifies polyadenylation sites (PAS) using first-strand reads and quantify its expression leveraging second-strand reads, consequently enabling detection of alternative PAS usage.
scTail embedded a pre-trained sequence model to remove the false positive clusters, which enabled us to further evaluate the reliability of the detection by examining the supervised performance metrics and learned sequence motifs.

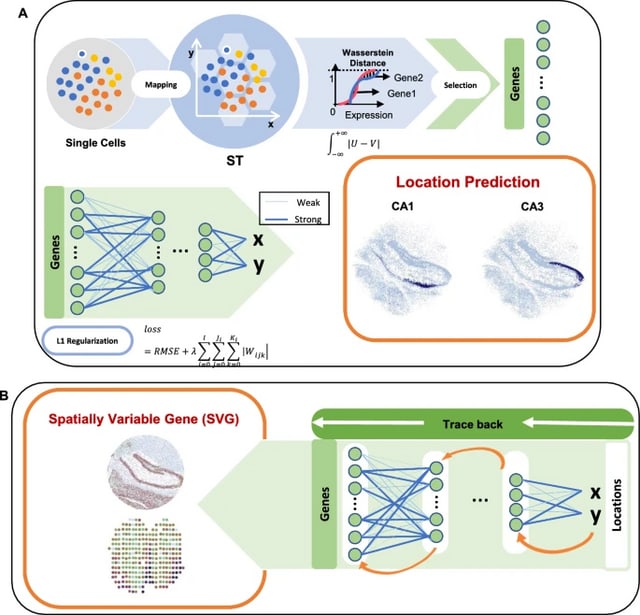
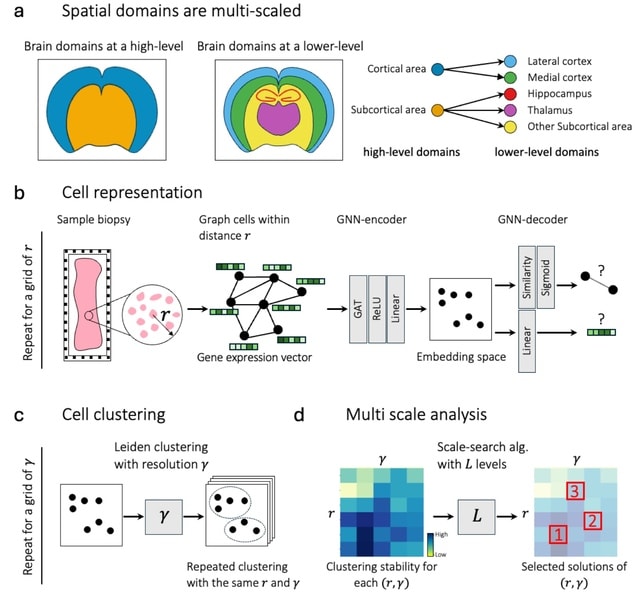
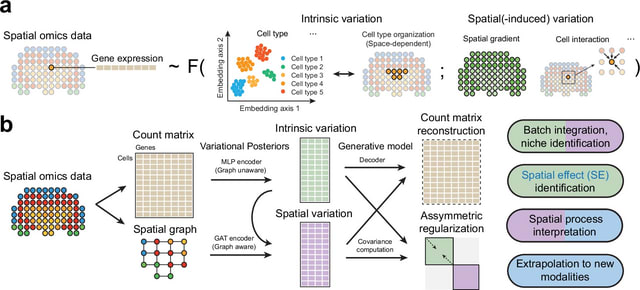
□ HarmoDecon: Mitigation of multi-scale biases in cell-type deconvolution for spatially resolved transcriptomics
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf451/8231072
HarmoDecon is a semi-supervised deep learning model that utilizes Gaussian Mixture Graph Convolutional Networks (GMGCN) architecture. It leverages the graph structure to update node features by message passing and assumes the node embeddings follow a Gaussian mixture model.
The rationale behind integrating GMGCN into HarmoDecon lies in its inherent ability to capture the spatial and gene expression similarities among SRT spots/pseudo-spots and reflect the fact that SRT spots are from different spatial domains.

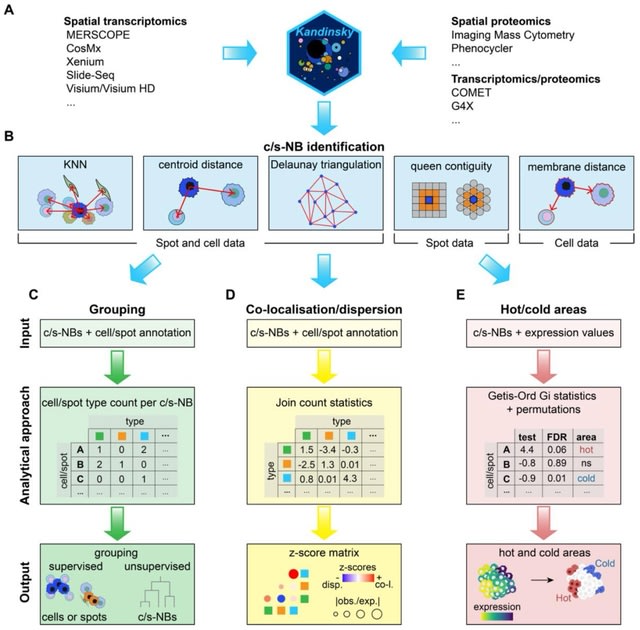
□ SpaFoundation: a visual foundation model for spatial transcriptomics
>> https://www.biorxiv.org/content/10.1101/2025.08.07.669202v1
SpaFoundation, a versatile visual foundational model with 80 million trainable parameters, pre-trained on 1.84 million histological image patches to learn general-purpose imaging representations.
SpaFoundation incorporated self-distillation and masked image modeling (MIM) to enhance the learning of high-level semantic and local structural features.

□ Predictive Gene Discovery with EPCY: A Density-Based Alternative to DE analysis
>> https://www.biorxiv.org/content/10.1101/2025.08.07.668357v1
EPCY, a method that ranks genes based on their predictive power using cross-validated classifiers and density estimation, without relying on null hypothesis testing.
EPCY employs a leave-one-out cross-validation scheme, training gene-specific Kernel Density Estimation (KDE) classifiers. EPCY directly assesses the overlap of expression profiles between groups using the MCC, offering a more balanced and less biased evaluation.

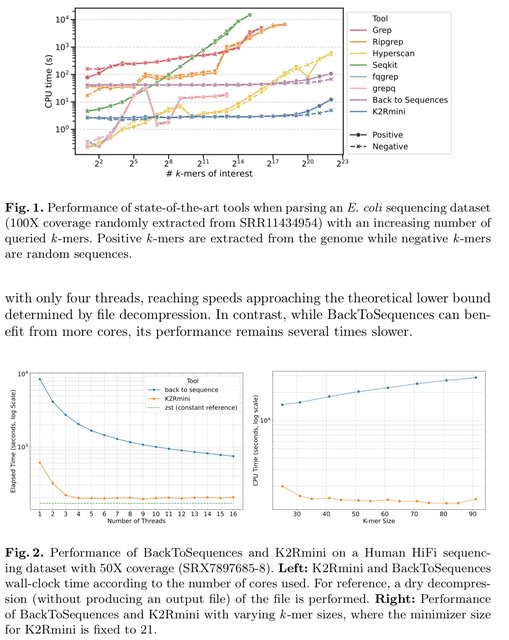
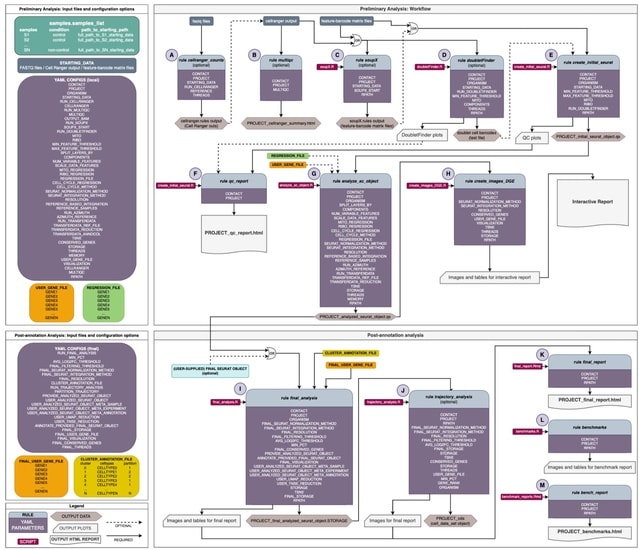
□ SingleRust: A High-Performance Toolkit for Single-Cell Data Analysis at Scale
>> https://www.biorxiv.org/content/10.1101/2025.08.04.668429v1
SingleRust is a computational framework for single-cell analysis that leverages systems programming principles. It is built on Rust’s ownership model and zero-copy semantics.
SingleRust reimplements six essential single-cell operations: quality control filtering, count normalization, highly variable gene identification, principal component analysis, differential expression testing, and k-nearest neighbor graph construction.

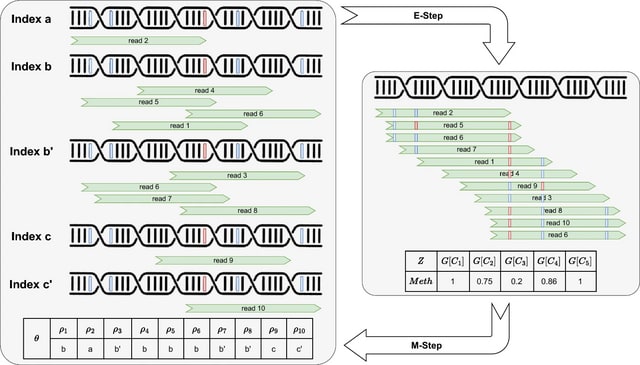
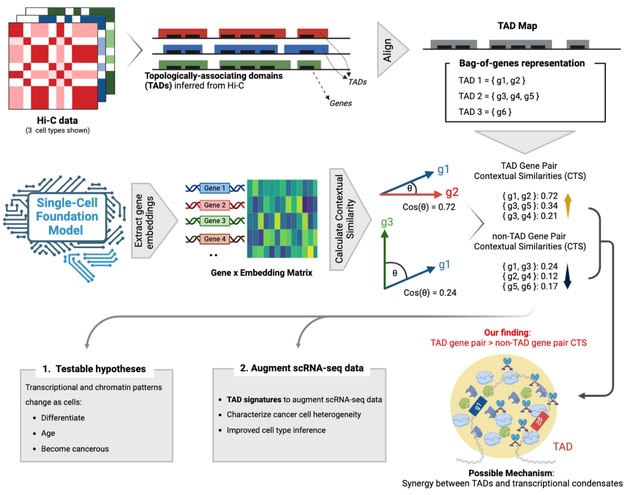
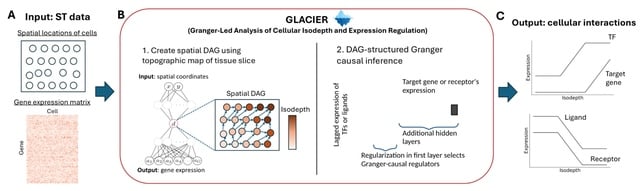
□ TENET: Tracing regulatory element networks using epigenetic traits to identify key transcription factors
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf435/8220914
TENET identifies key transcription factors (TFs) and regulatory elements (REs) linked to a specific cell type by detecting correlations between gene expression and RE methylation in case–control datasets, and identifying top genes by number of RE methylation site links.
TENET utilizes DNA methylation and gene expression datasets from any cell or disease group to identify key TEs and REs. All of TENET's functions, including those for searching TFs, using topologically associating domains to further characterize the target genes of REs and TFs.

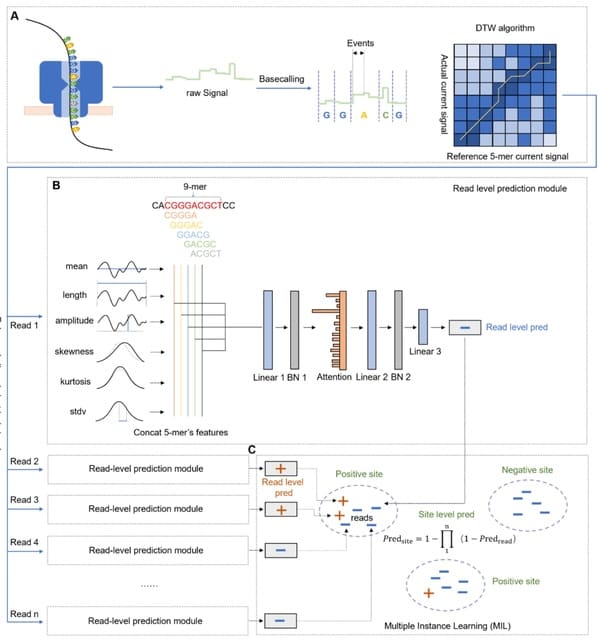
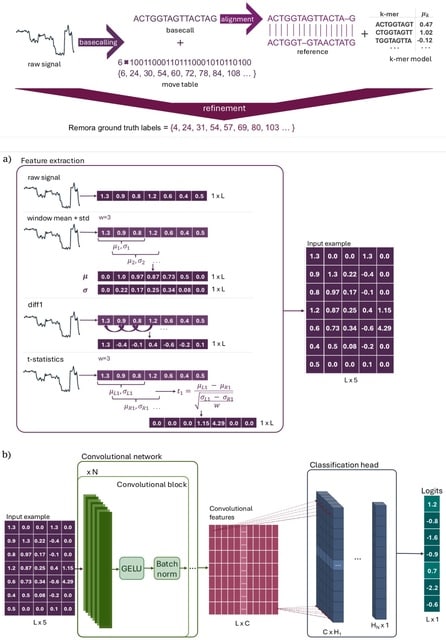
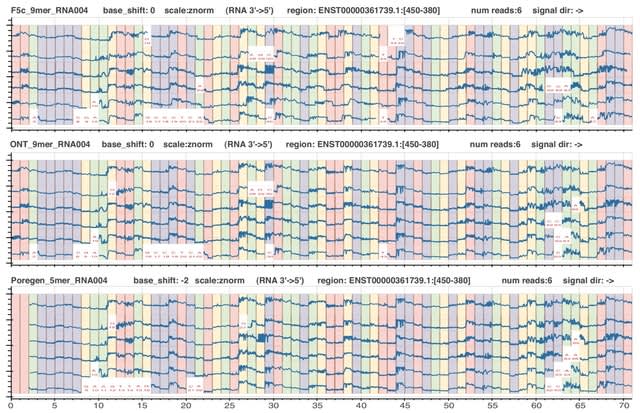
□ MultiNano: Accurate detection and quantification of single-base m6A RNA modification using nanopore signals with multi-view deep learning
>> https://www.biorxiv.org/content/10.1101/2025.08.04.668591v1
MultiNano, a multi-view learning model that integrates raw signal and basecalling features. This integration enables a more comprehensive and accurate characterization of m6A modification distribution across multiple species.
The MultiNano framework are composed of three main components: the data preprocessing module, the MultiNano core module, and the classification module. Initially, Nanopore DRS reads are processed to extract relevant features.
Basecalling features are fed into a BiLSTM module to capture sequential dependencies, while raw signal features transformed into Gramian Angular Summation Field (GASF) representations, and raw signals were processed through a 1D residual networks (ResNet) module.
These representations are then further analyzed by an optimized ResNet2D module. This module enhances spatial feature extraction performance by combining channel-wise attention (via SE blocks) and spatial attention mechanisms.
Finally, all features were fused through a fully connected layer. The classification module then employed a multiple instance learning (MIL) strategy to aggregate read-level methylation probabilities and infered site-level m6A modification probabilities.

□ scGCM: Semi-supervised contrastive learning variational autoencoder Integrating single-cell multimodal mosaic datasets
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06239-5
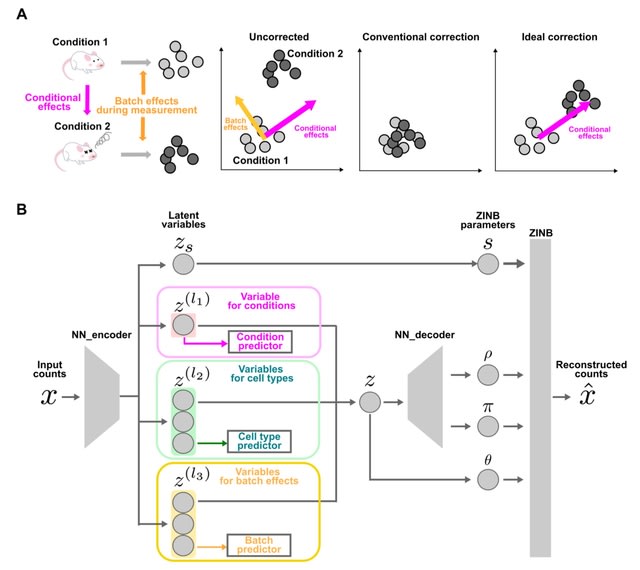
scGCM(single-cell Graph Contrastive Modular variational autoencoder) integrates single-cell multimodal mosaic data and eliminate batch effects. It represents single-cell data as graph structures and utilizes graph structures to preserve both local and global features of cells.
scGCM maintains the topological structure of the data during dimensionality reduction. scGCM employs neighborhood graphs and contrastive learning to effectively eliminate batch effects, ensuring robust integration of different modalities within the embedded space.

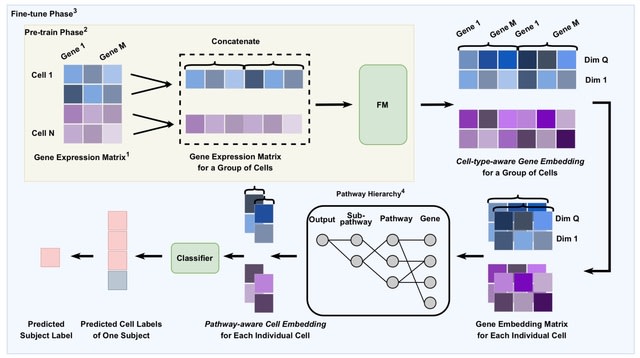
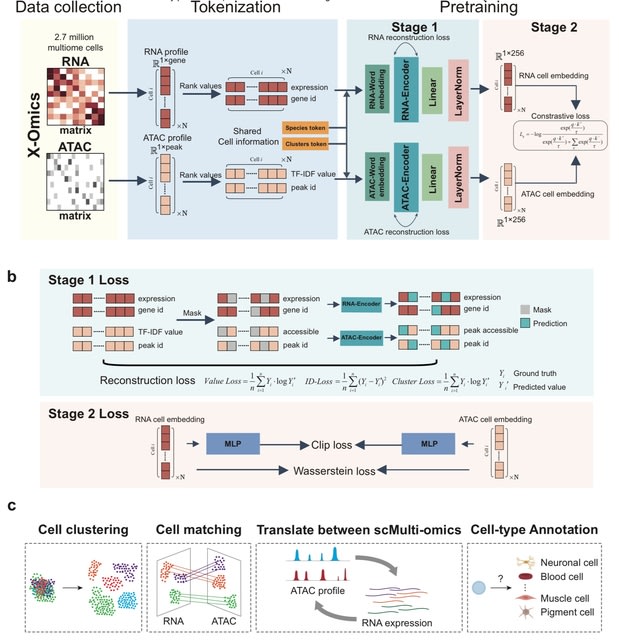
□ scMomer: A modality-aware pretraining framework for single-cell multi-omics modeling under missing modality conditions
>> https://www.biorxiv.org/content/10.1101/2025.08.04.668374v1
scMomer, a modality-aware pretraining framework designed for multi-modal representation learning under missing modality conditions. scMomer adopts a three-stage pretraining strategy that learns unimodal cell representations, models joint representations from multi-omics data.
scMomer distills multi-modal knowledge to enable multi-omics-like representations from unimodal input. Its modality-specific architecture and three-stage pretraining strategy enable effective learning under missing modality conditions and help capture cellular heterogeneity.

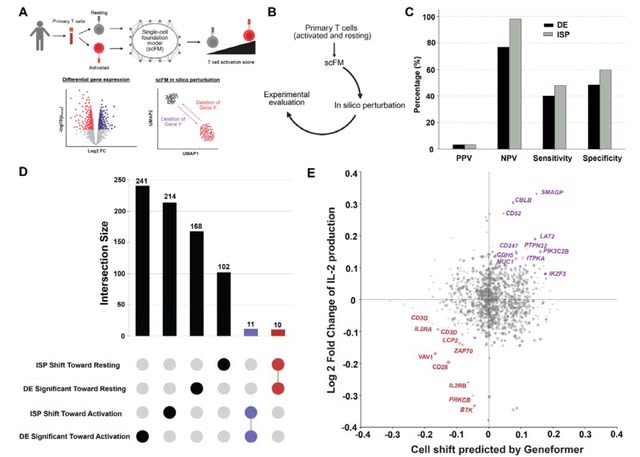
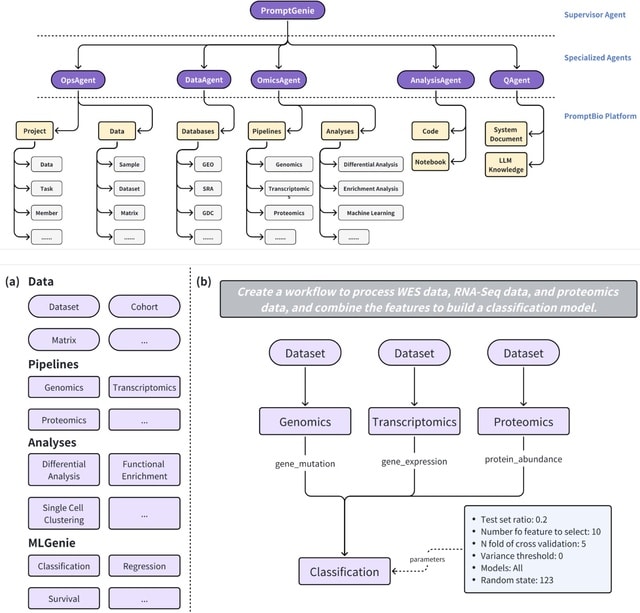
□ OmniCellAgent: Towards AI Co-Scientists for Scientific Discovery in Precision Medicine
>> https://www.biorxiv.org/content/10.1101/2025.07.31.667797v1
OmniCellAgent empowers non-computational-expert users-such as patients and family members, clinicians, and wet-lab researchers-to conduct scRNA-seq data-driven biomedical research like experts, uncovering molecular disease mechanisms and identifying effective precision therapies.
OmniCellTOSG (Omni-Cell Text-Omic Signaling Graph) is a large-scale, graph-structured, Al-ready dataset that harmonizes single-cell transcriptomics data and biological knowledge graph.
The graph structure of OmniCellTOSG encodes both molecular attributes (e.g., gene expression profiles, pathway activities) and biological relationships (e.g., signaling pathways and protein-protein interactions), allowing intelligent agents to reason over complex omic landscapes.

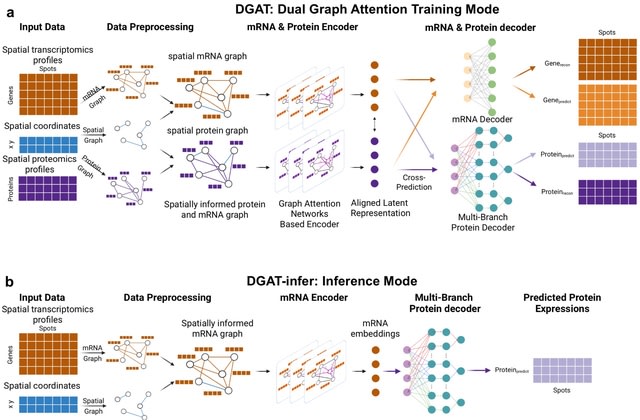
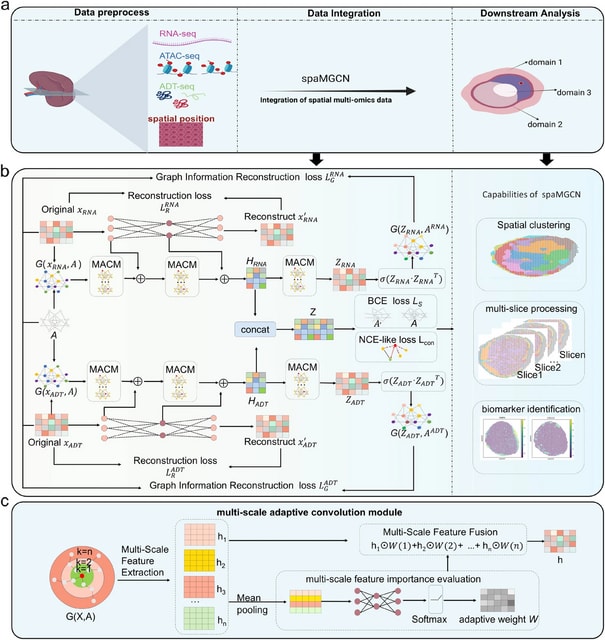
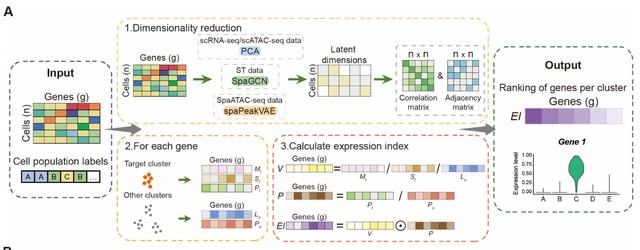
□ SpaMV: Interpretable spatial multi-omics data integration and dimension reduction
>> https://www.biorxiv.org/content/10.1101/2025.08.02.668264v1
Spatial Multi-View representation learning (SpaMV), a novel spatial multi-omics integration algorithm designed to explicitly disentangle cross-modal shared features and modality-specific private features into distinct latent spaces.
SpaMV minimizes mutual information between the inferred private latent variable from one modality and data from other modalities, preventing leakage of shared information into private latent spaces. It incorporates a non-parametric test to enforce statistical independence.

□ scDIAGRAM: Detecting Chromatin Compartments from Individual Single-Cell Hi-C Matrix without Imputation or Reference Features
>> https://www.biorxiv.org/content/10.1101/2025.08.01.668129v1
scDIAGRAM (single-cell compartments annotation by Direct stAtistical modeling and GRAph coMmunity detection), a novel computational tool designed to annotate chromatin A/B compartments in scHi-C data.
scDIAGRAM takes an intrachromosomal Hi-C contact matrix as input, dividing the genome into discrete regions at specified resolution. It performs 2D change-point detection followed by graph partitioning to mitigate inherent noise in data and annotate compartments for each locus.

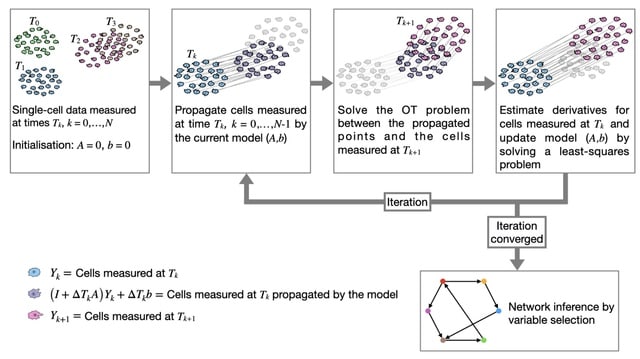
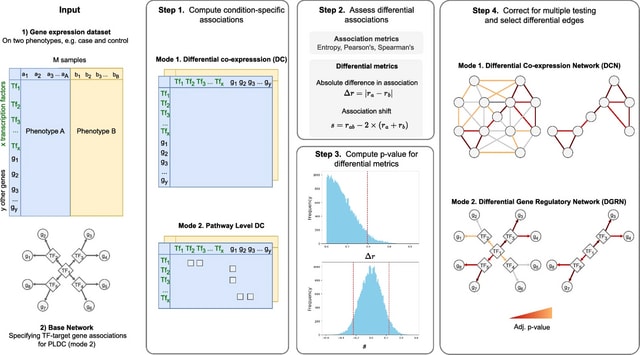
□ Double Optimal Transport for Differential Gene Regulatory Network Inference with Unpaired Samples
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf352/8221768
Double OT conceptualizes changes in gene expression between states as a mass transport problem and proposes a two-level Optimal Transport framework to infer large-scale differential GRNs for paired or unpaired samples.
Double OT determines edge scores by solving the robust OT problem and handles unpaired samples by incorporating a partial OT-based sample alignment step. Double OT explicitly models gene regulation as a mass transportation problem from the perspective of OT theory.

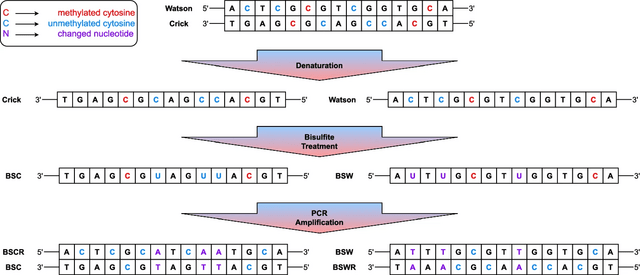
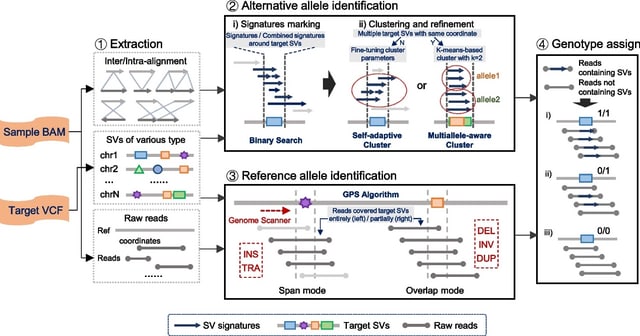
□ Snappy: de novo identification of DNA methylation sites based on Oxford Nanopore reads
>> https://www.biorxiv.org/content/10.1101/2025.08.03.668330v1
Snappy combines motif enrichment with simultaneous analysis of basecalling results. Snappy is primarily oriented on Oxford Nanopore data, but unlike Snapper, it does not use any heuristics, does not require control sample sequencing, and is significantly easier to run.

□ GenomicLayers: sequence-based simulation of epi-genomes
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06224-y
GenomicLayers, a new R package to run rules-based simulations of epigenetic state changes genome-wide in Eukaryotes. GenomicLayers enables scientists working on diverse eukaryotic organisms to test models of gene regulation in silico.

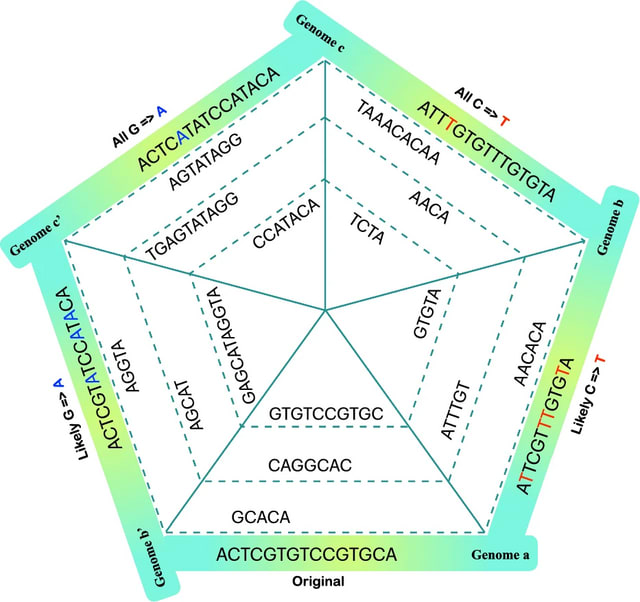
□ Dna-storalator: a computational simulator for DNA data storage
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06222-0
The DNA-Storalator is a cross-platform software tool that simulates in a simplified digital point of view biological and computational processes involved in the process of storing data in DNA molecules.
The simulator receives an input file with the designed DNA strands that store digital data and emulates the different biological and algorithmical components of DNA-based storage system.
The DNA-Storalator adopts an abstracted error model that captures key error characteristics while enabling high adaptability. It can incorporate factors such as GC-dependent error rates and error-prone motifs, tailoring the model to different synthesis or sequencing conditions.


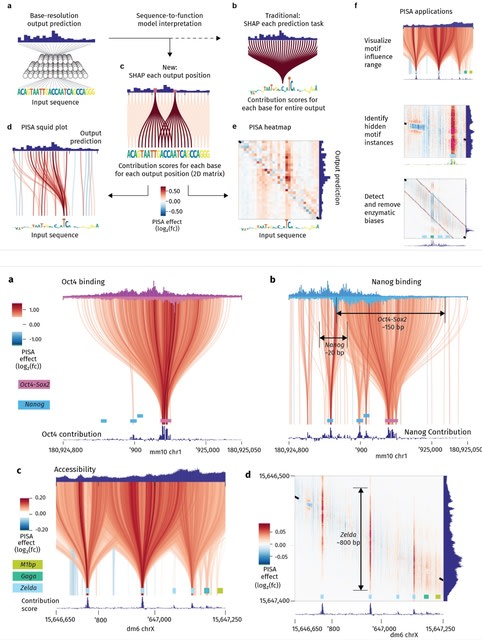
□ tangermeme: A toolkit for understanding cis-regulatory logic using deep learning models
>> https://www.biorxiv.org/content/10.1101/2025.08.08.669296v1
tangermeme implements "everything-but-the-model" when it comes to genomic deep learning. tangermeme is intentional, as the computational layers w/in the models and their training strategies are much more rapidly evolving than the ways in which these models are subsequently used.


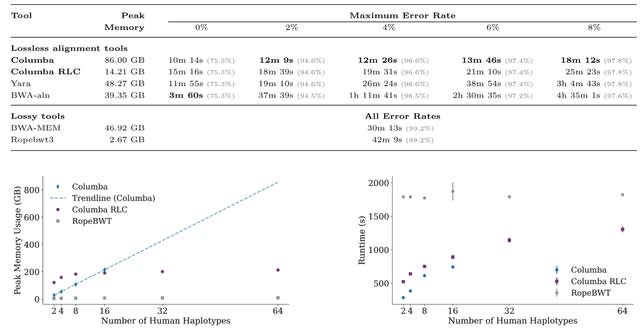
□ NOVOLoci: Unlocking the full potential of Oxford Nanopore reads
>> https://www.biorxiv.org/content/10.1101/2025.08.08.669243v1
NOVOLoci, a haplotype-aware assembler capable of high-quality targeted and whole-genome assemblies, despite the relatively high error rates of Oxford Nanopore Technologies data.
By adopting a novel seed-extension approach with iterative conflict resolution, it achieves accurate haplotype phasing, thus overcoming a critical limitation of current graph-based assemblers.
NOVOLoci outperforms the 4 leading assembly tools across 5 clinically relevant genomic disorder loci by delivering accurately phased assemblies w/ superior contiguity and completeness, even compared w/ hybrid assemblers - nearly triple the N90 value compared w/ Verkko hybrid.

□ Hi-Enhancer: a two-stage framework for prediction and localization of enhancers based on Blending-KAN and Stacking-Auto models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf441/8232719
Hi-Enhancer employs a Blending-KAN model, which integrates the results of various base classifiers and employs Kolmogorov-Arnold Networks (KAN) as a meta-classifier to predict enhancers based on flexible combinations of multiple epigenetic signals.
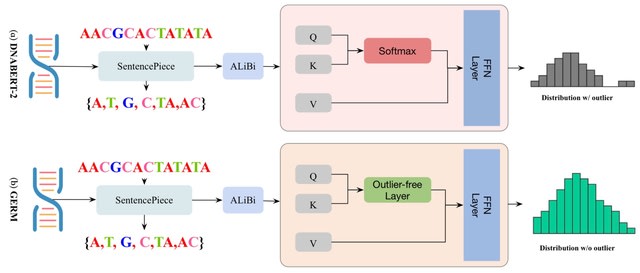
Hi-Enhancer uses a Stacking-Auto model, which extracted sequence features using DNABERT-2 and located the enhancers based on the Stacking strategy and AutoGluon framework. Hi-Enhancer utilizes a dynamic thresholding algorithm to pinpoint the complete boundaries of enhancers.

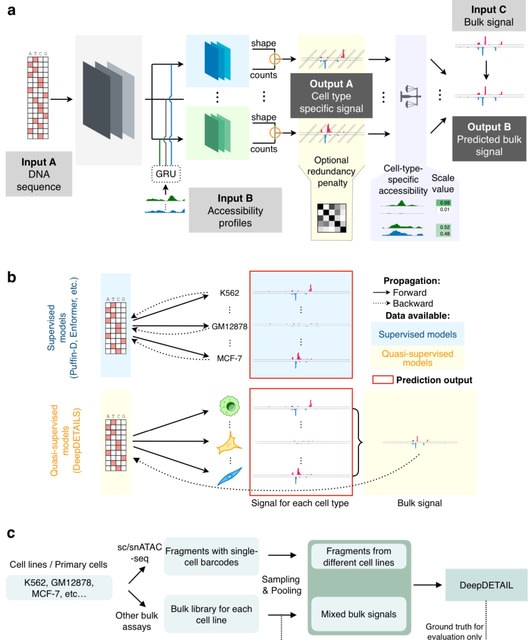
□ CLM-access: A Specialized Foundation Model for High-dimensional Single-cell ATAC-seq analysis https://www.biorxiv.org/content/10.1101/2025.08.10.669570v1
CLM-access - a Transformer-based cell language foundation model designed for scATAC-seq data. To handle the high dimensionality, CLM-access partitions accessible chromatin regions into patches, each consisting of a fixed number of peaks, and treated each patch as a token.
CLM-access inputs combine token embeddings with peak-level representations and are processed through a Transformer architecture to perform masked peak reconstruction, optimized using binary cross-entropy (BCE) loss.




































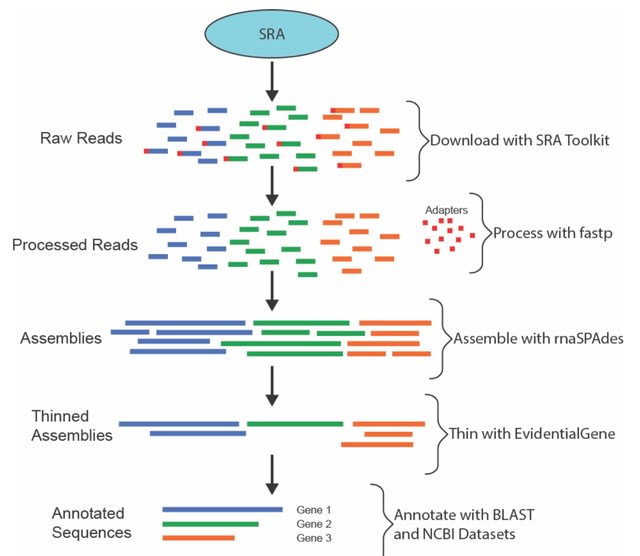
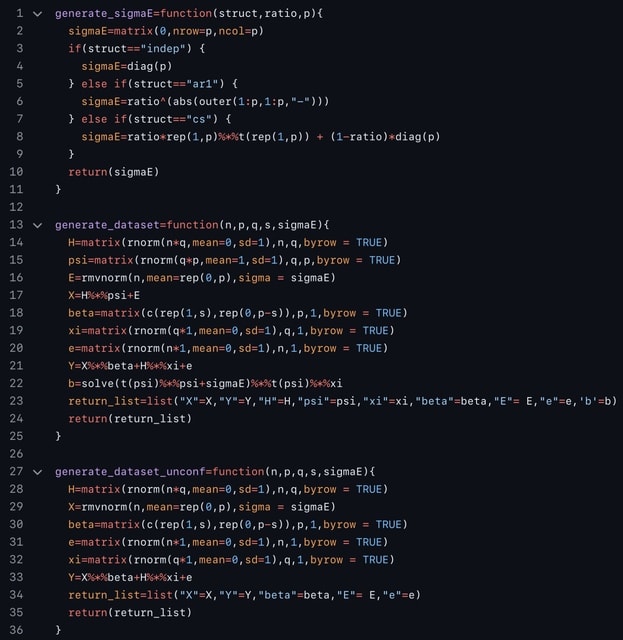
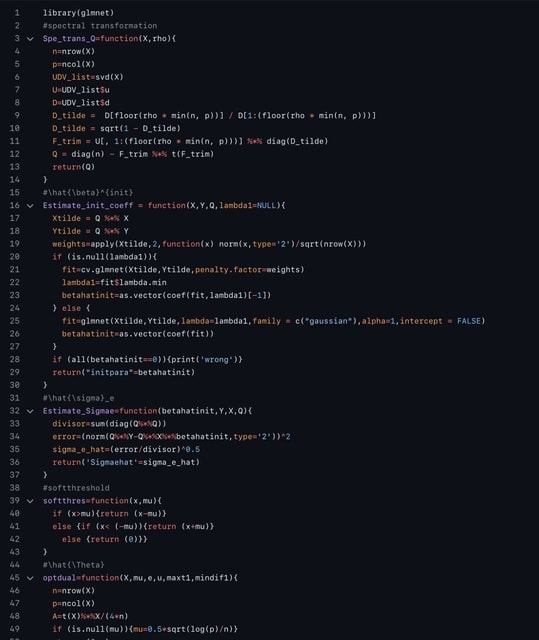
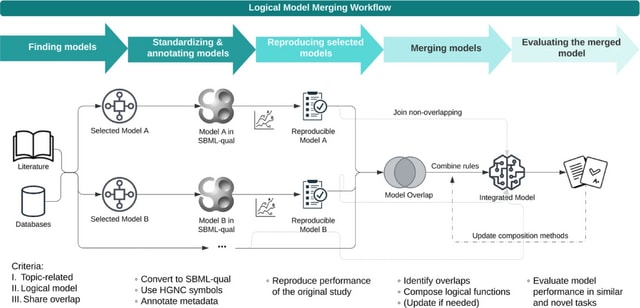
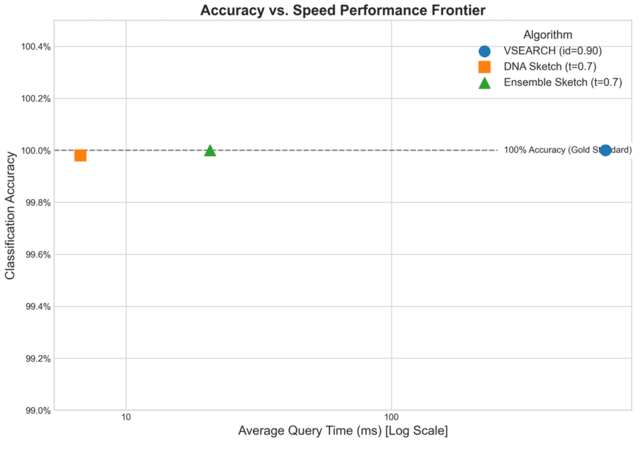
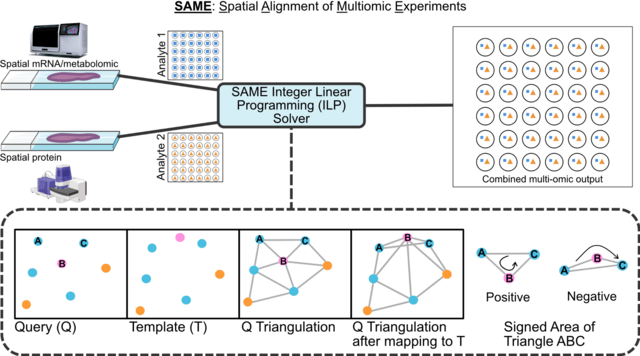

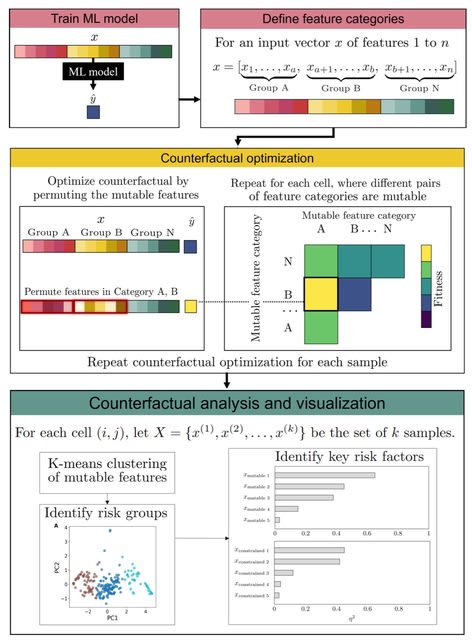

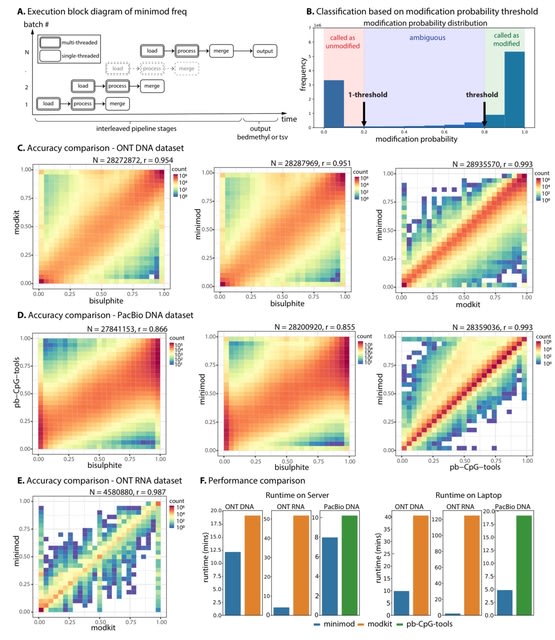












































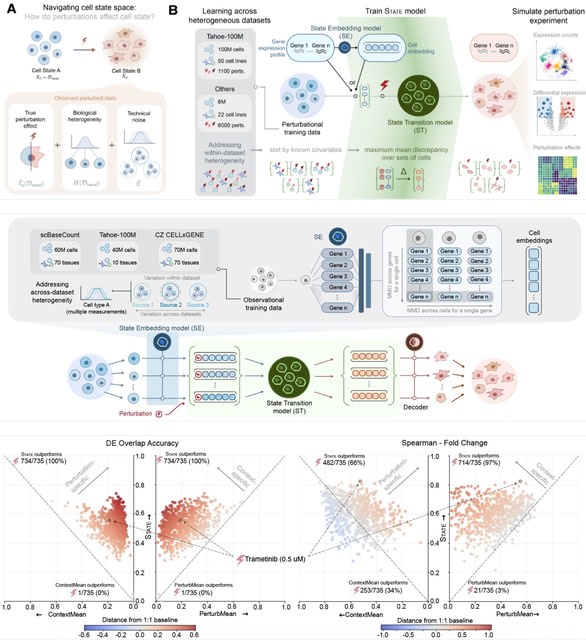
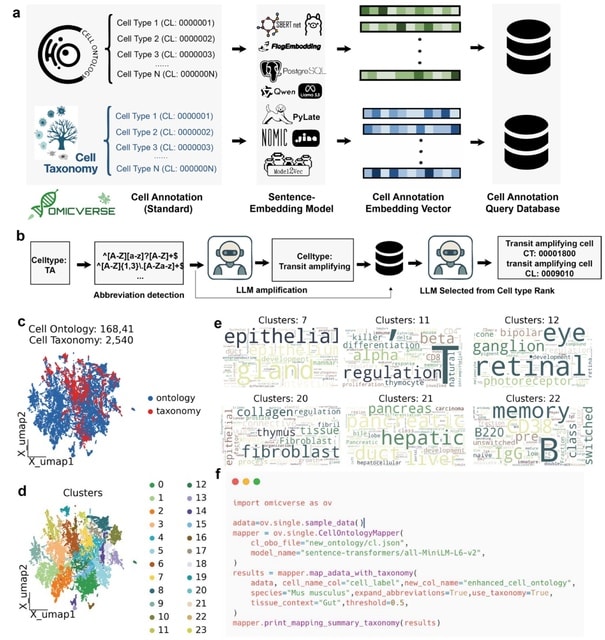
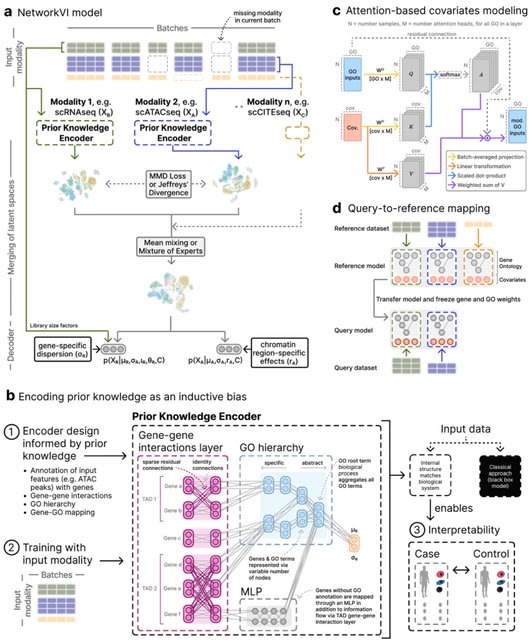
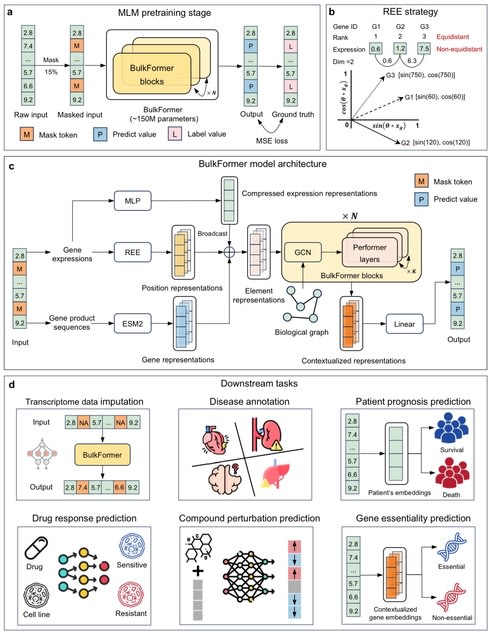


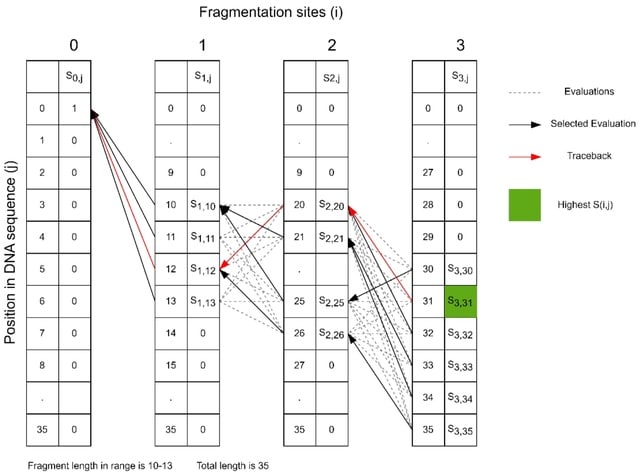
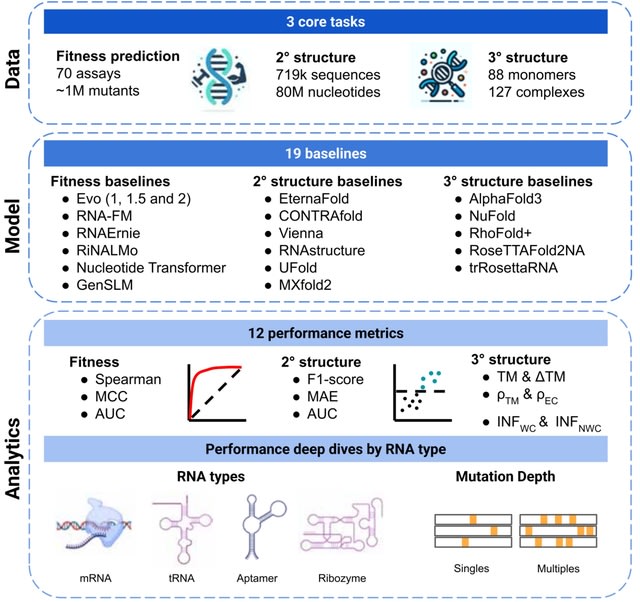
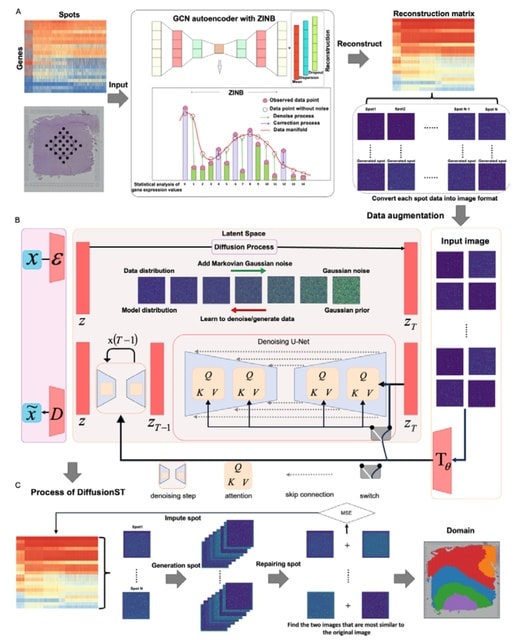

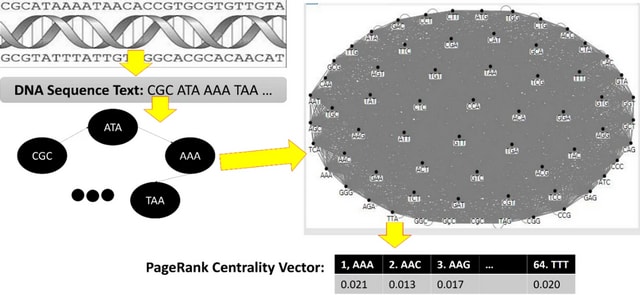


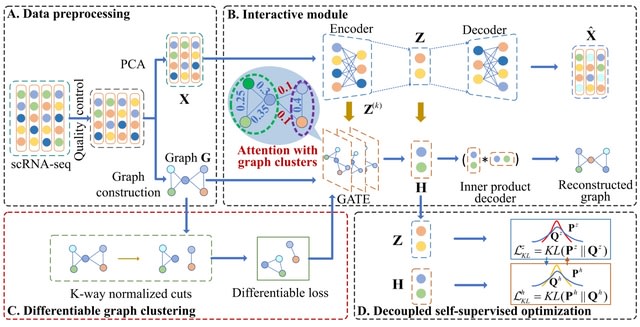
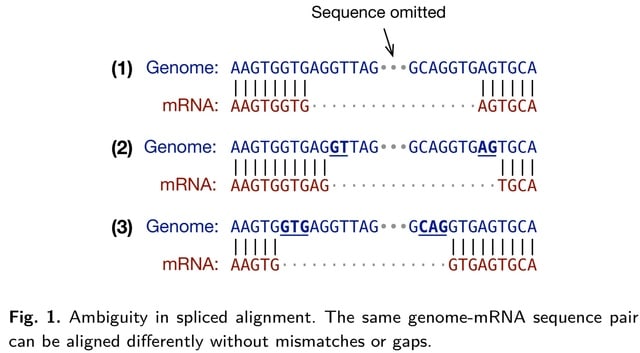
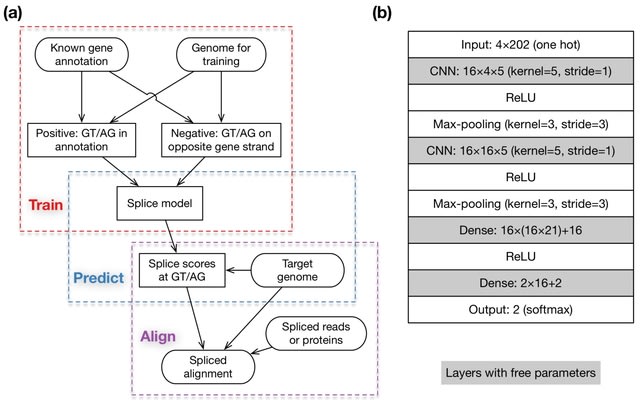
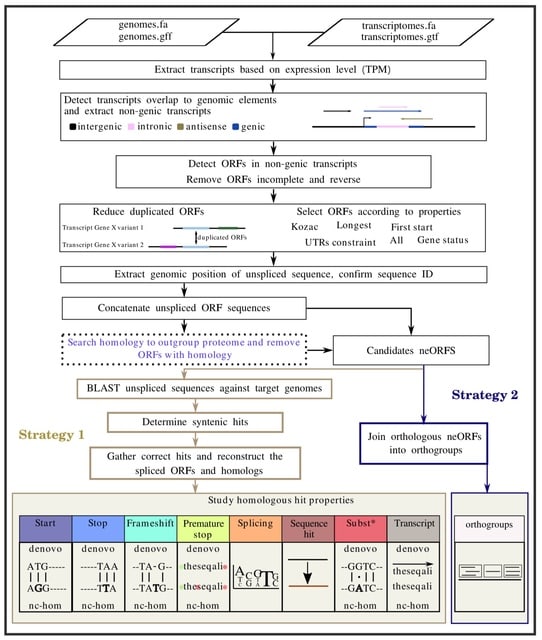


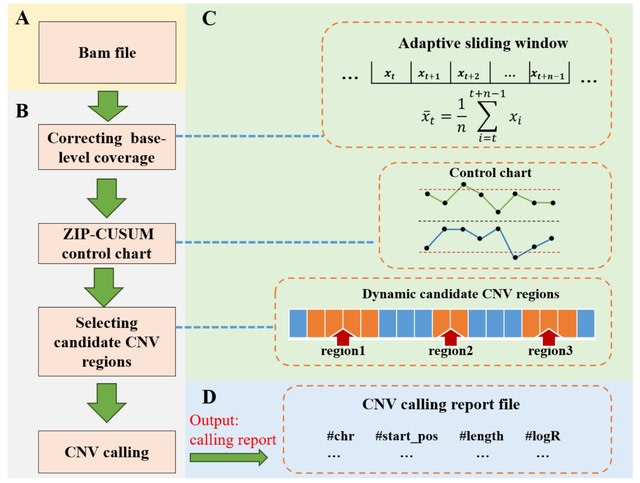

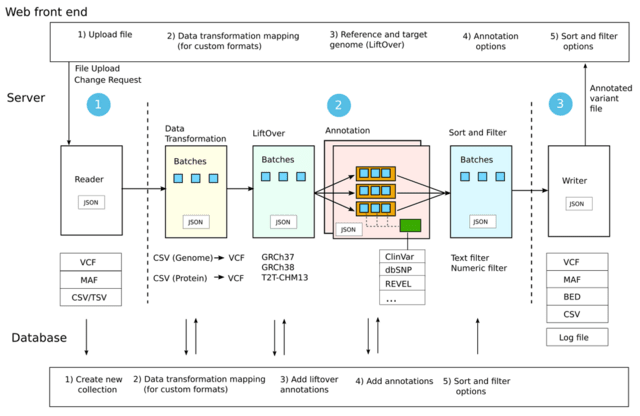
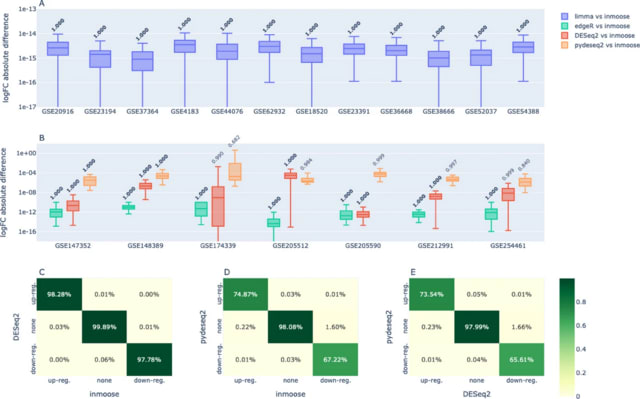
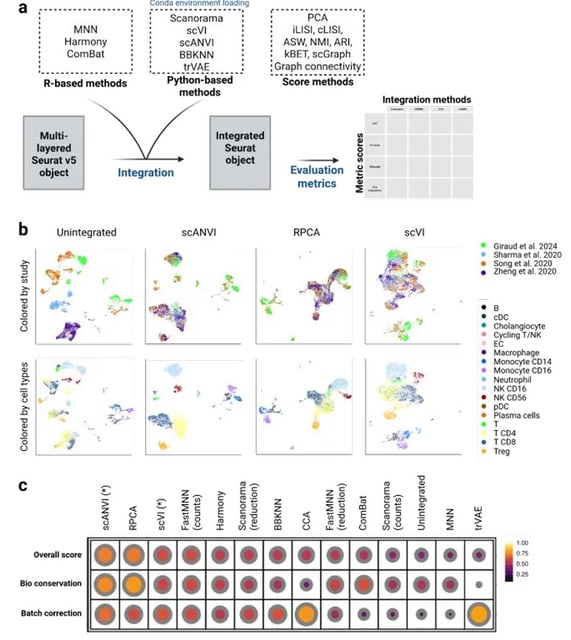






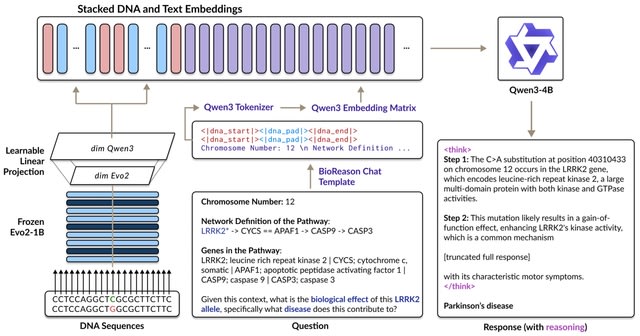

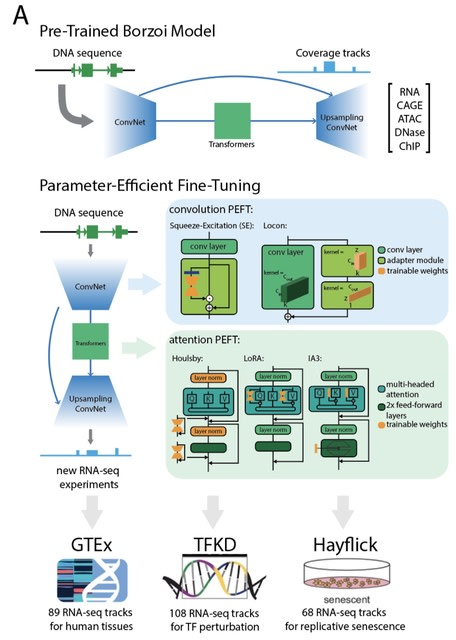

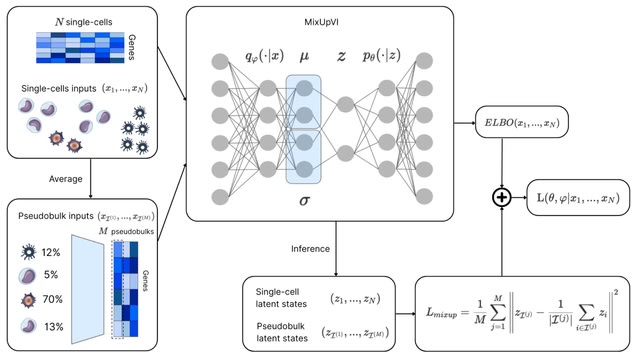


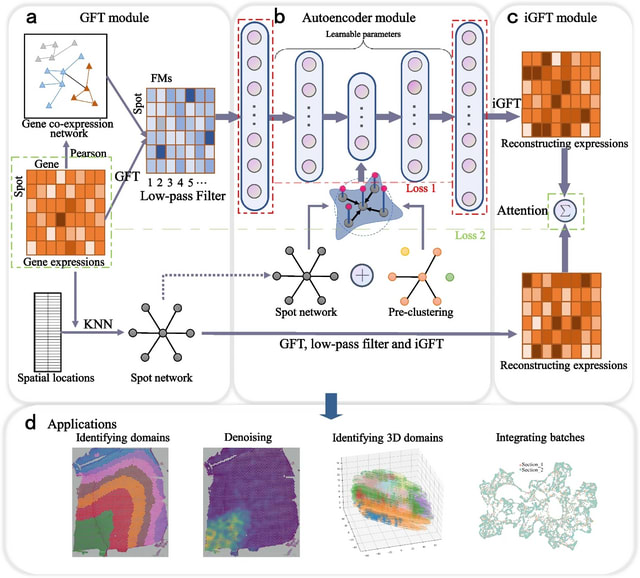

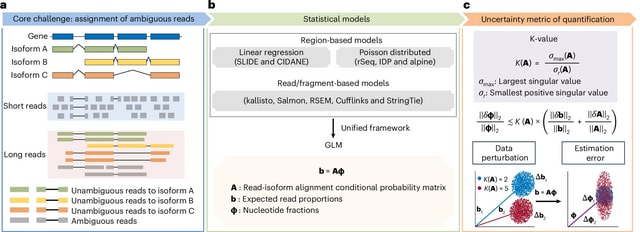


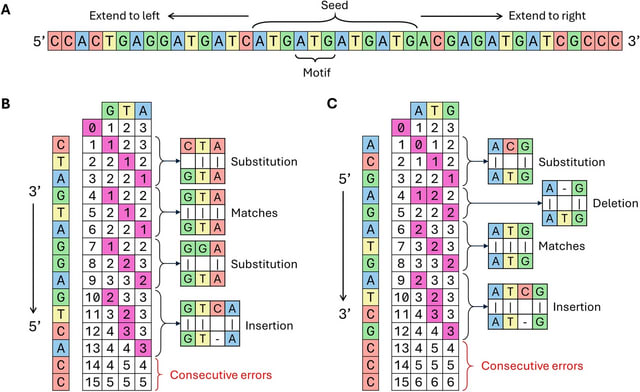

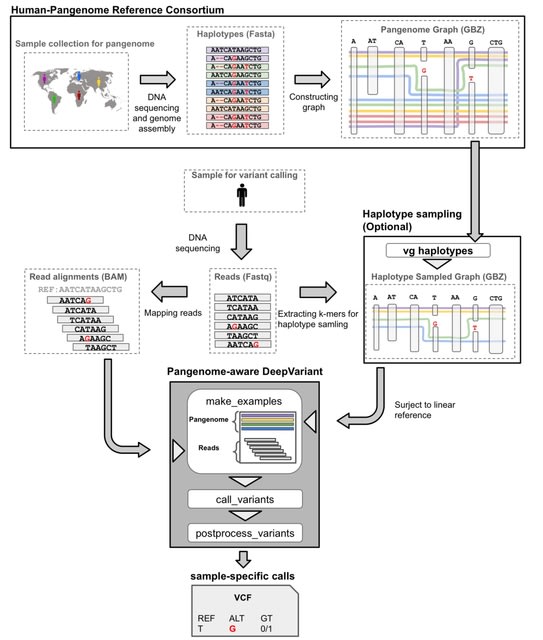


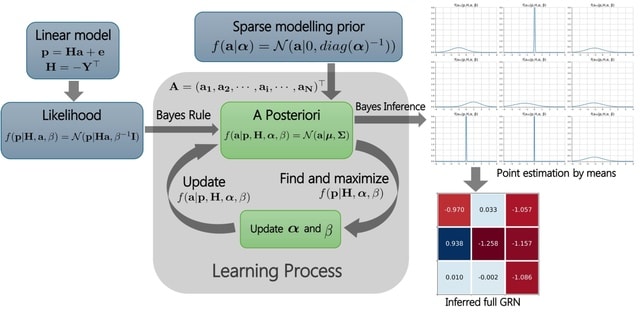
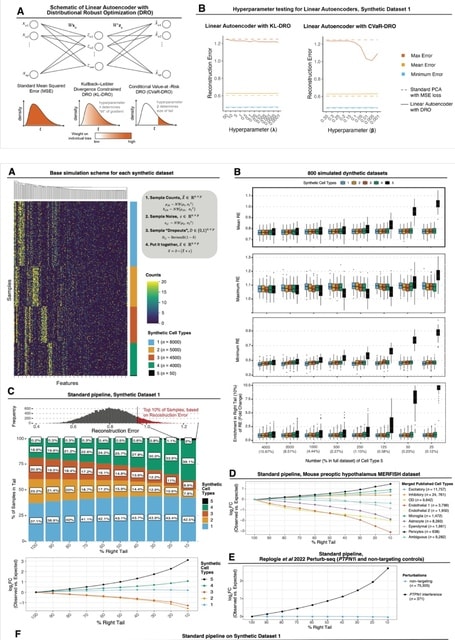


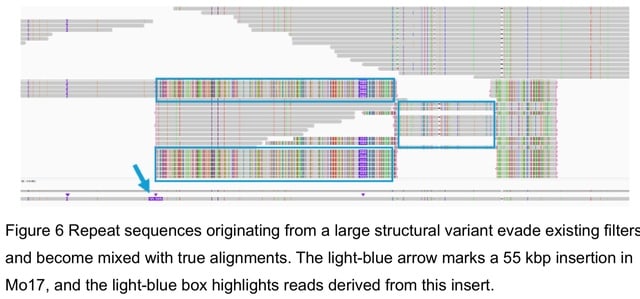
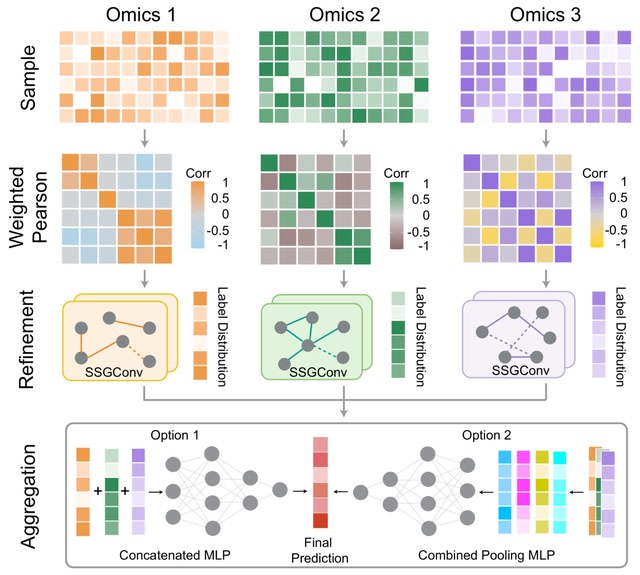
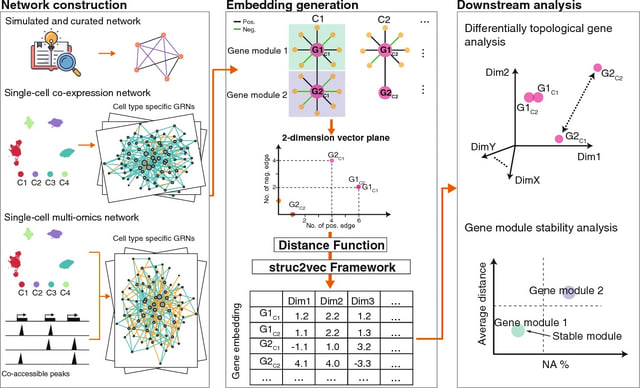
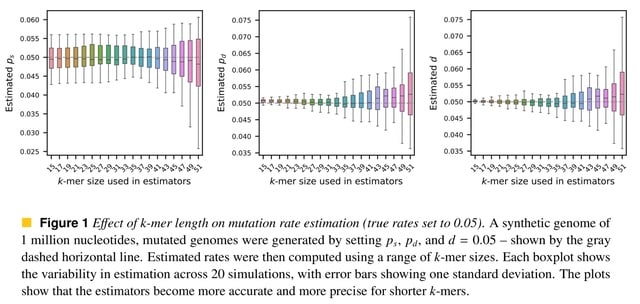
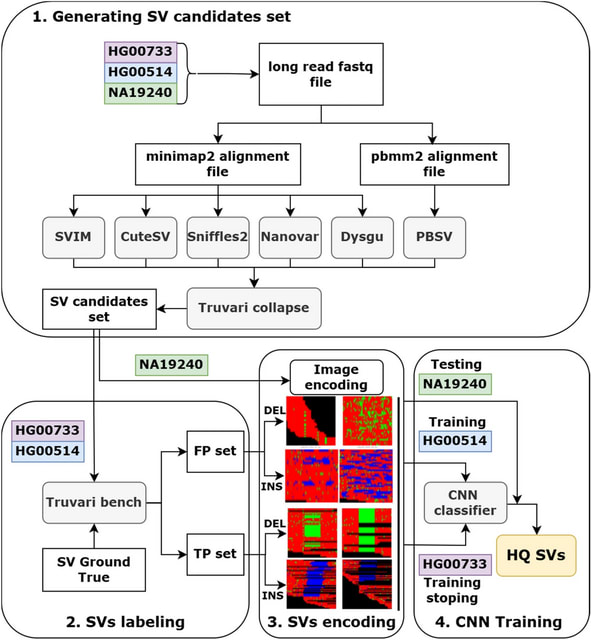

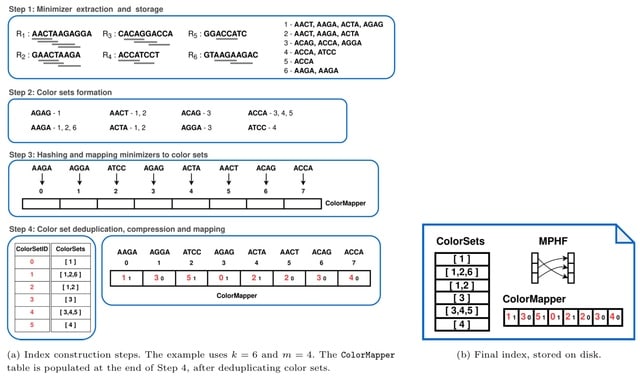
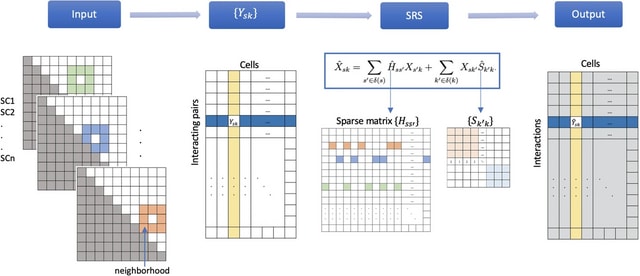
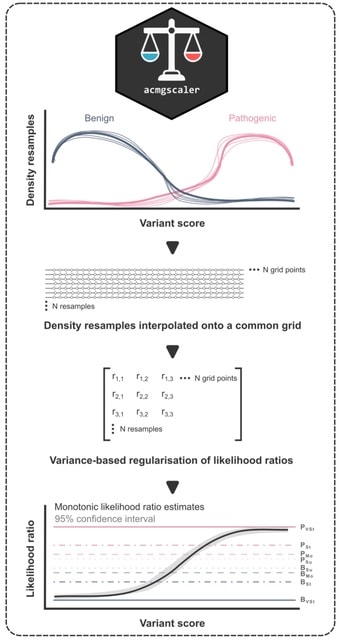
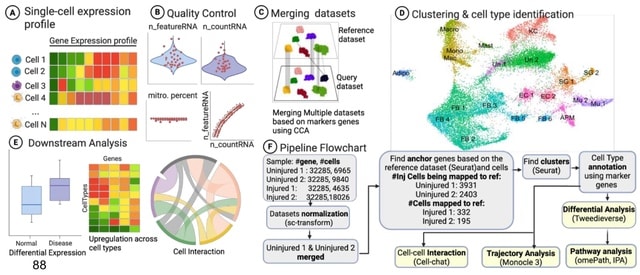

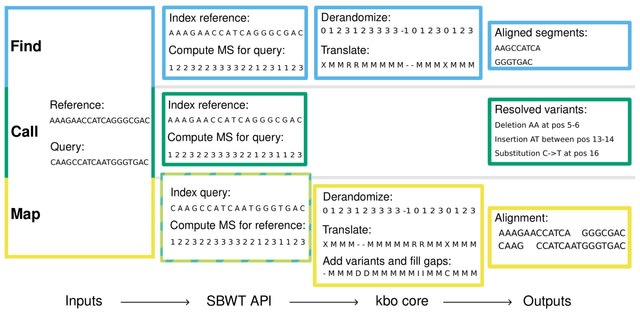
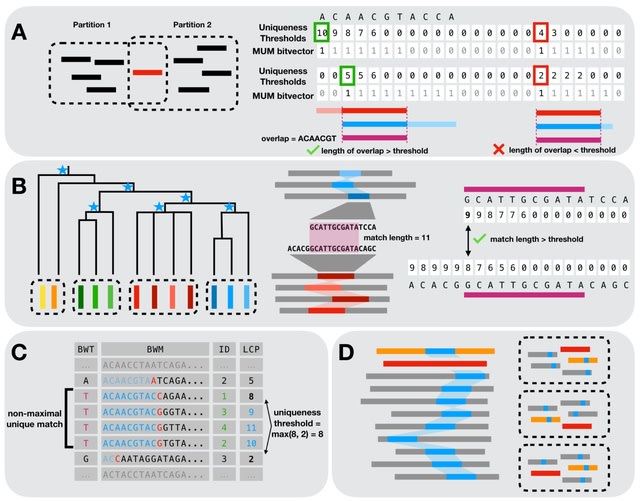
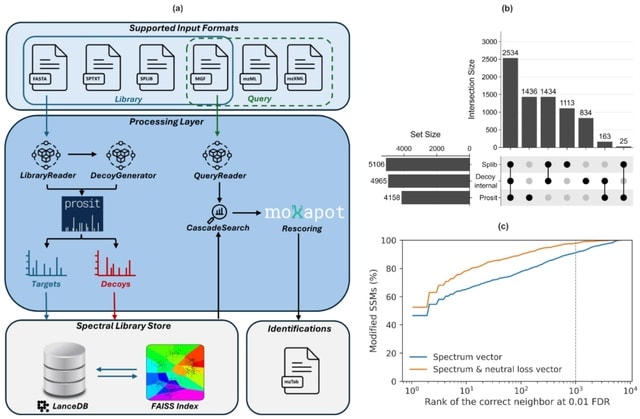
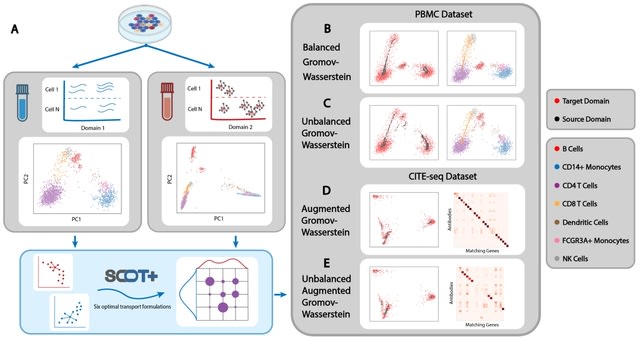




















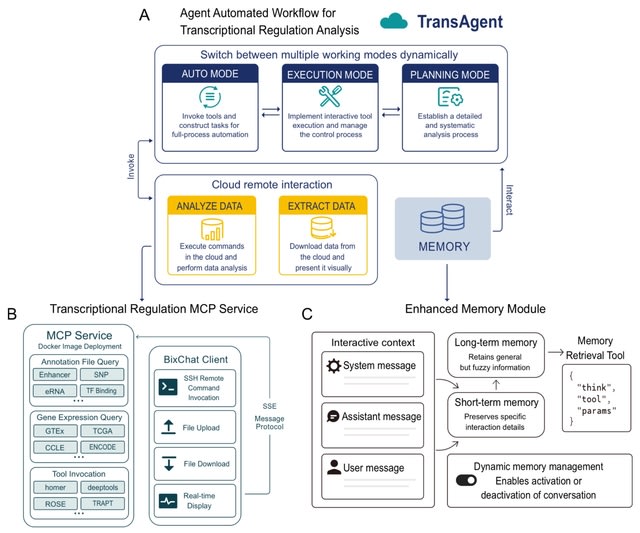

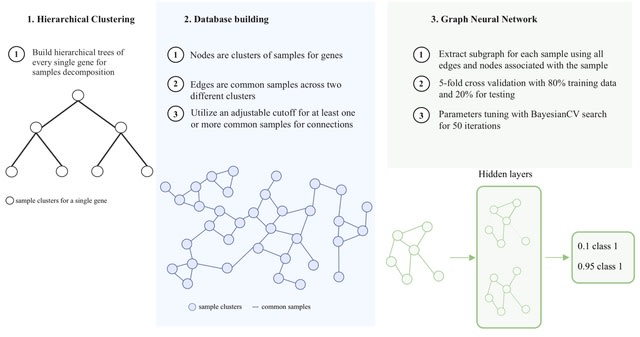




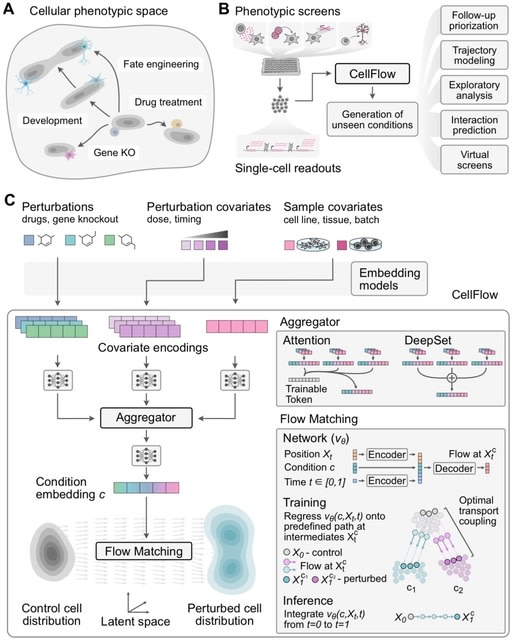
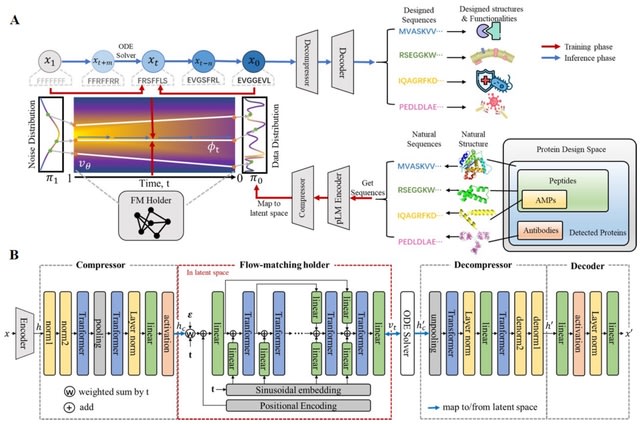








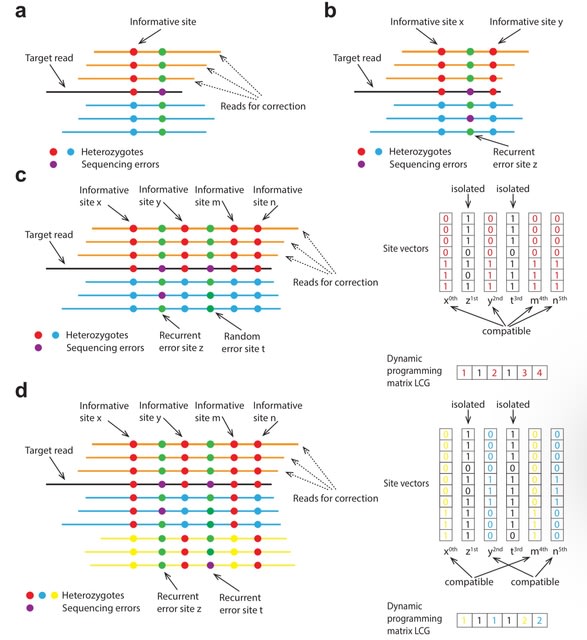

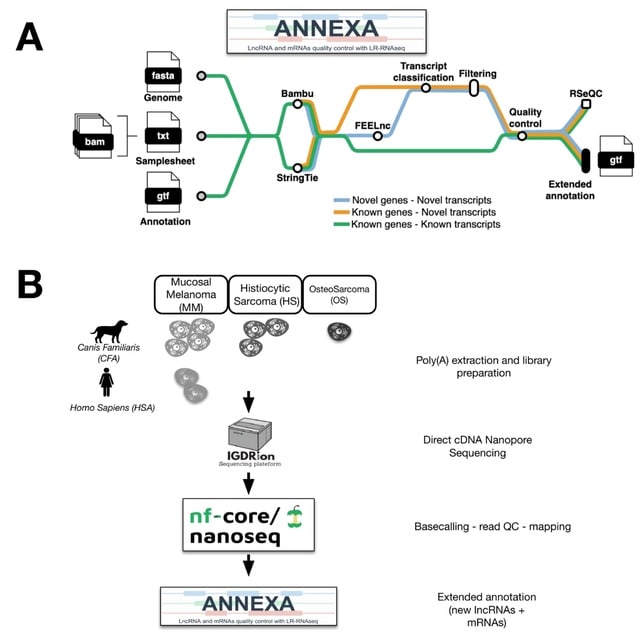

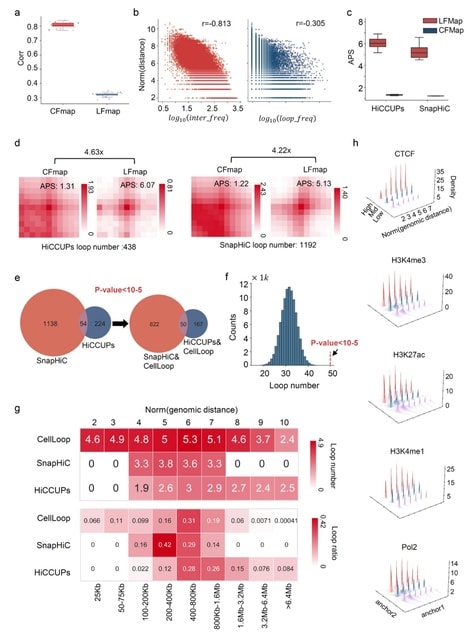

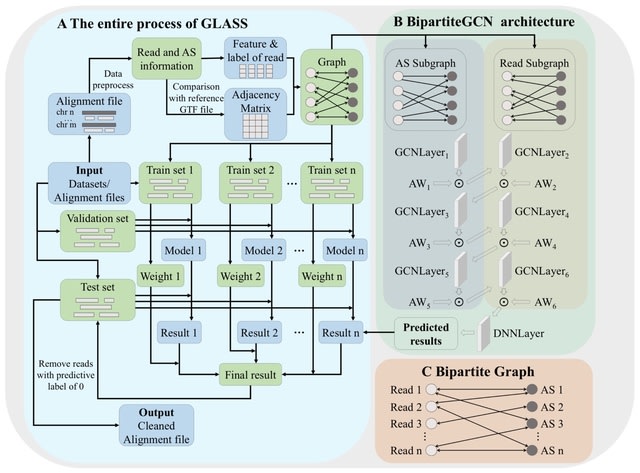
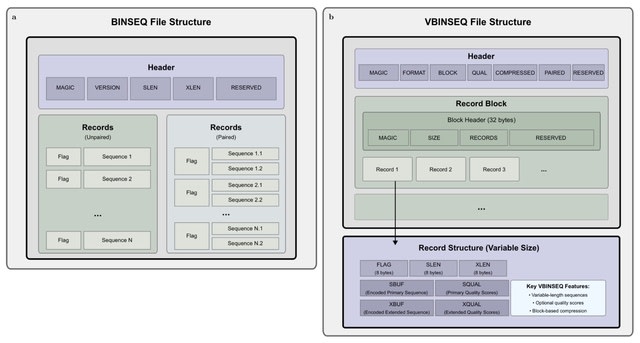
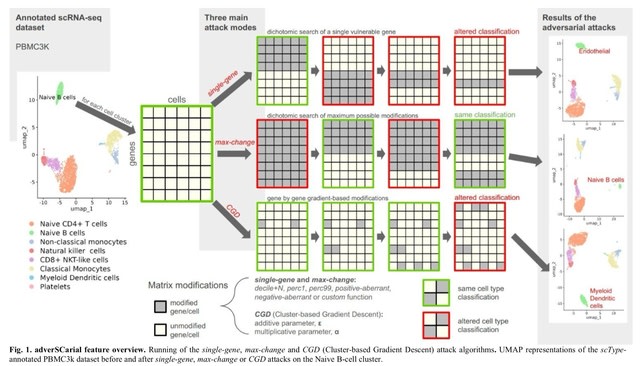
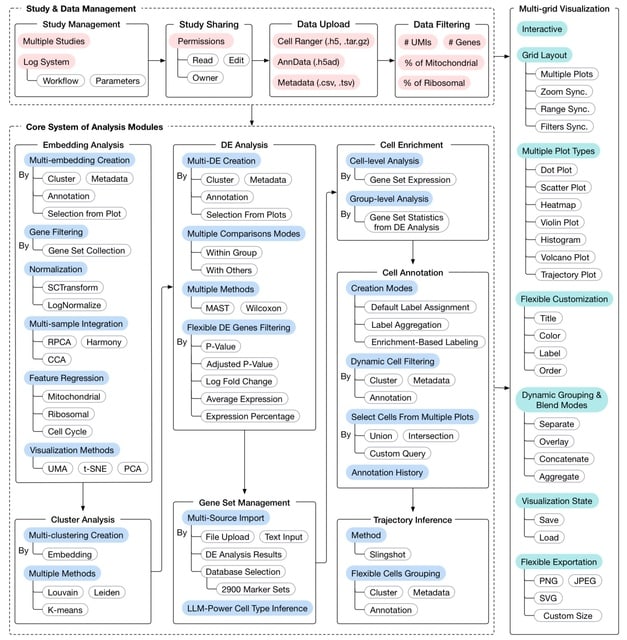
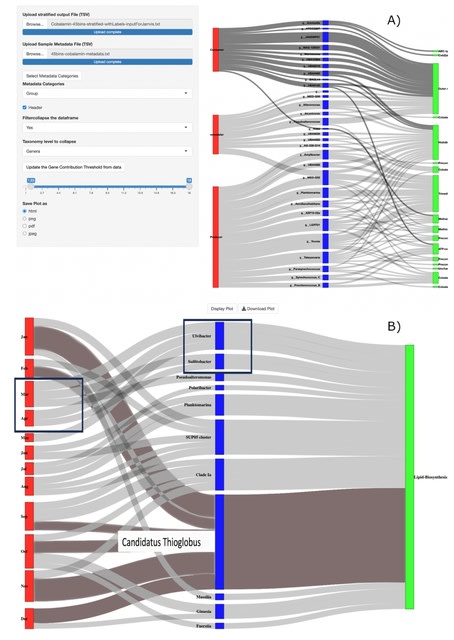




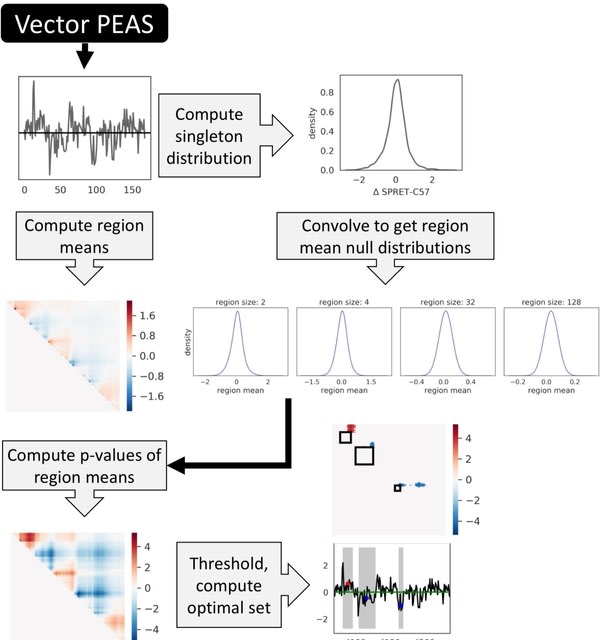
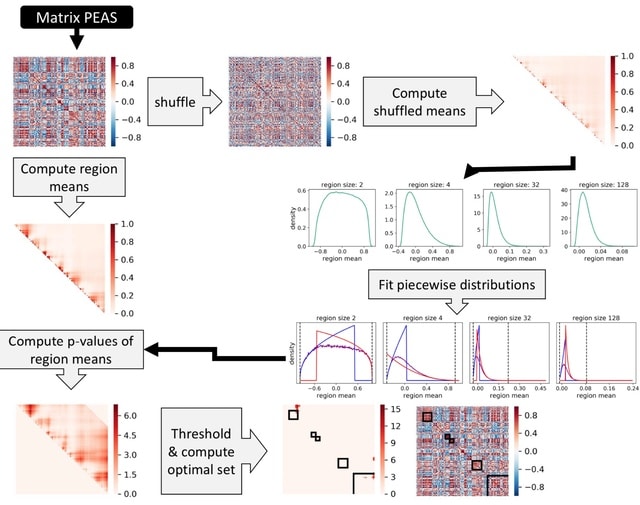


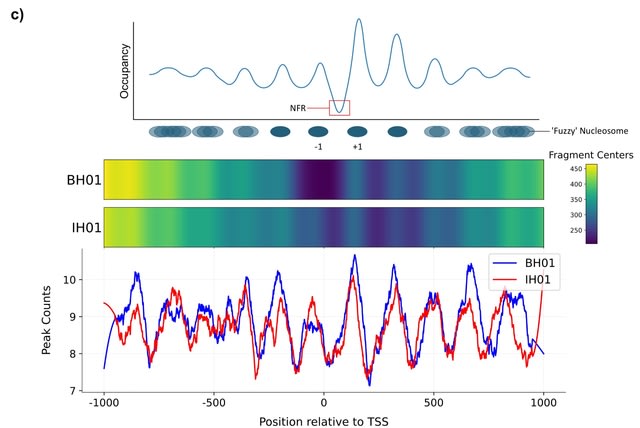
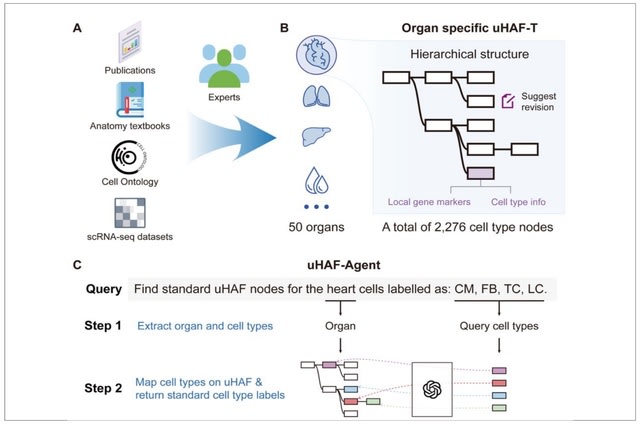


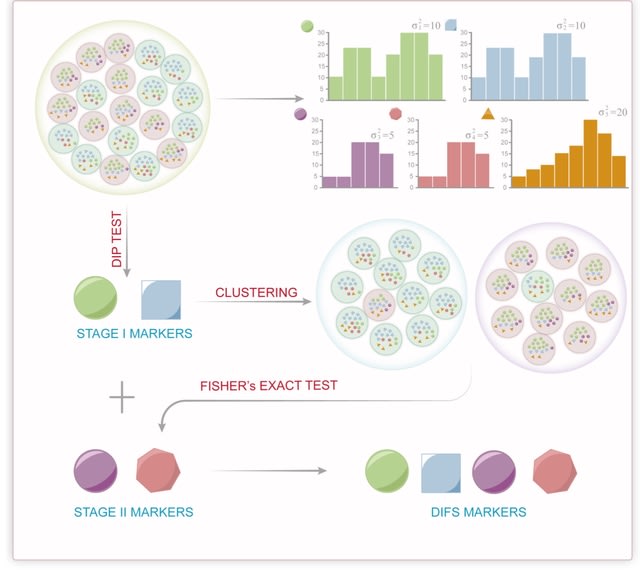
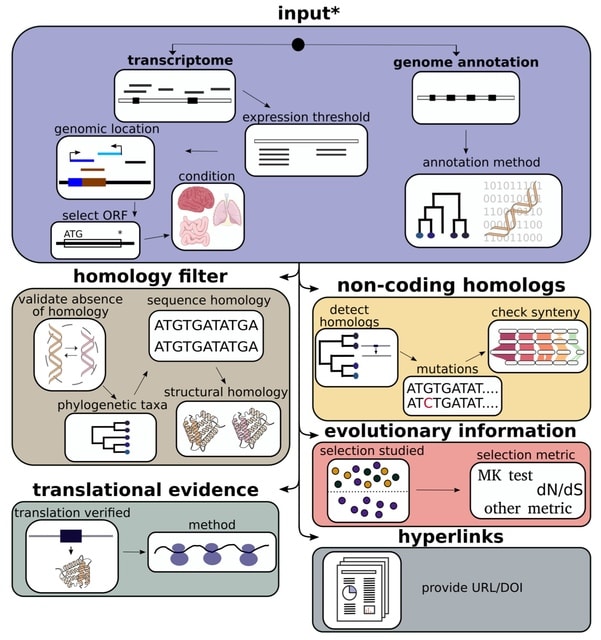
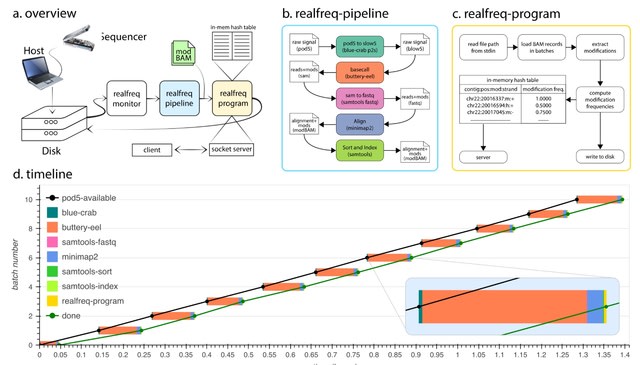
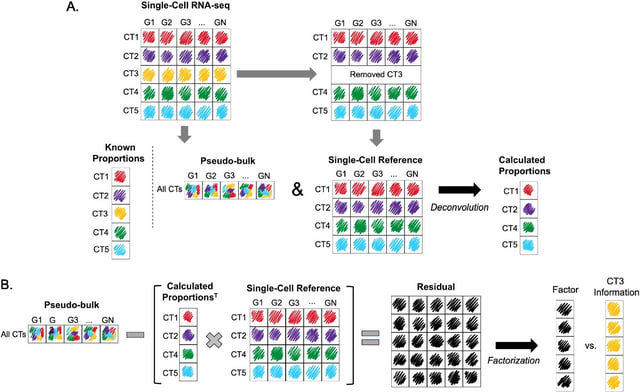
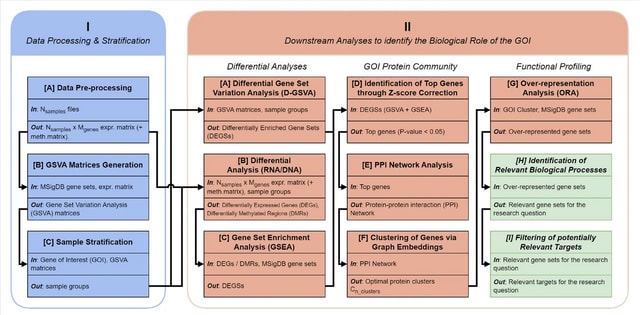
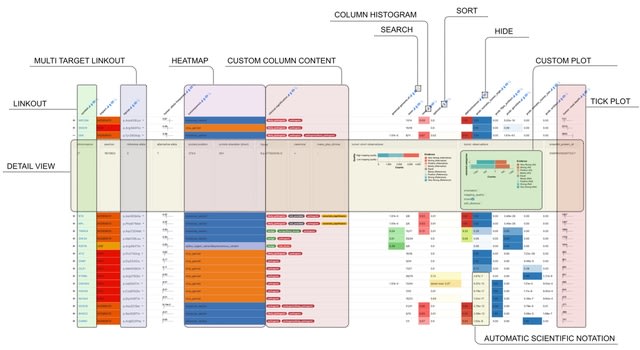
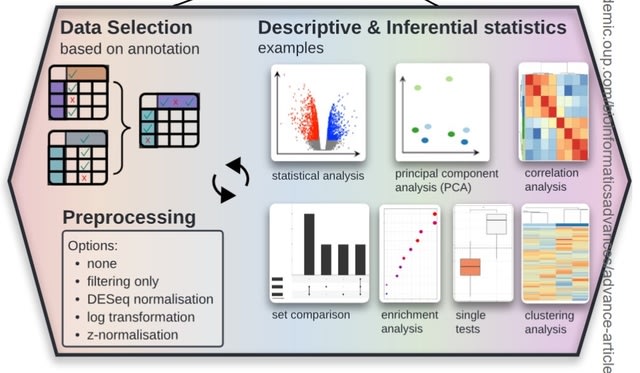

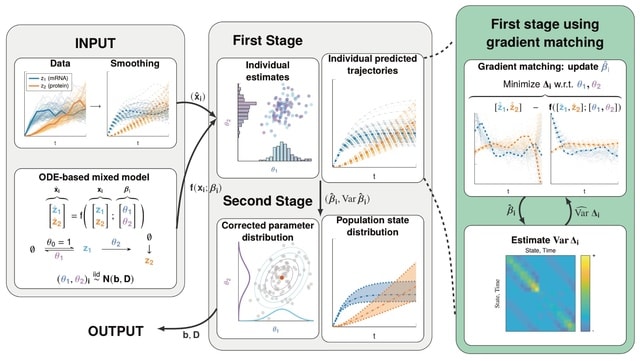

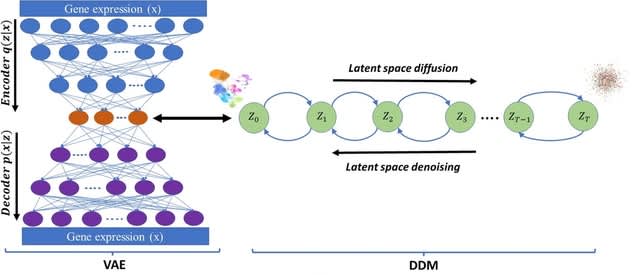
 □ MNMO: Discover driver genes from a Multi-Omics data based multi-layer network
□ MNMO: Discover driver genes from a Multi-Omics data based multi-layer network 












 (Created with Midjourney v6.1)
(Created with Midjourney v6.1)