
□ ApexOracle: Predicting and generating antibiotics against future pathogens
>>
https://arxiv.org/abs/2507.07862
ApexOracle integrates three foundational representation modules. The genomic encoder employs Evo2, a DNA language model pretrained on genomes spanning all domains of life, to transform a pathogen's entire genome into a numerical representation that captures genotypic hallmarks.
ApexOracle incorporates pathogen-specific context through the integration of molecular features. captured via a foundational discrete diffusion language model-and a dual-embedding framework that combines genomic- and literature-derived strain representations.

□ Tranquillyzer: A Flexible Neural Network Framework for Structural Annotation and Demultiplexing of Long-Read Transcriptomes
>>
https://www.biorxiv.org/content/10.1101/2025.07.25.666829v1
Tranquillyzer (TRANscript QUantification In Long reads-anaLYZER) employs a hybrid neural network architecture that integrates convolutional neural networks to detect local sequence motifs with BiLSTM layers to model long-range dependencies across the read.
Tranquillyzer supports an alternate model variant incorporating a conditional random field layer, enforcing structured transitions between predicted labels. It allows precise classification even with noncanonical configurations, shortened motifs, or internal structural artifacts.

□ Decipher: Joint representation and visualization of derailed cell states
>>
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03682-8
Decipher (deep characterization of phenotypic derailment) is an interpretable deep generative model for the simultaneous integration and visualization of gene expression and cell state from normal and perturbed single-cell RNA-seq data, revealing shared and disrupted dynamics.
Decipher uses linear transformations / single-layer neural networks to connect all representations w/n a unified probabilistic framework, flexible enough to learn nonlinear mechanisms while imposing a rigid inductive bias that prevents arbitrary distortion of the global geometry.
Decipher components represent the dominant axes of variation:progression/derailment. It learns the dependency structure of cell-state latent factors w/ the top latent space embedding, enabling the discovery of both shared and unique biological mechanisms from sparse trajectories.
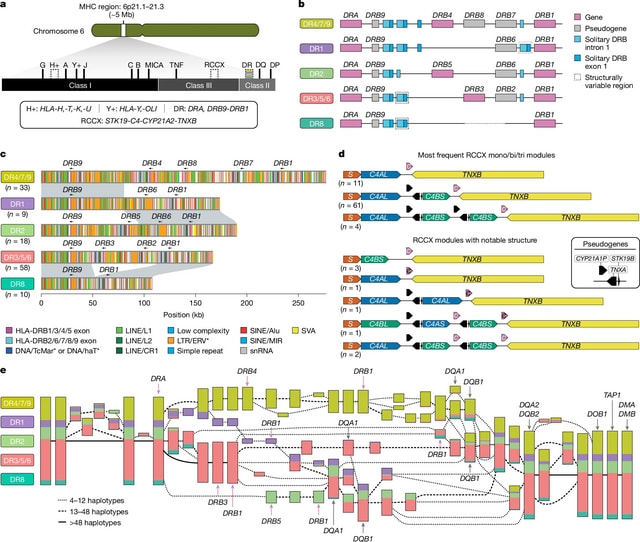
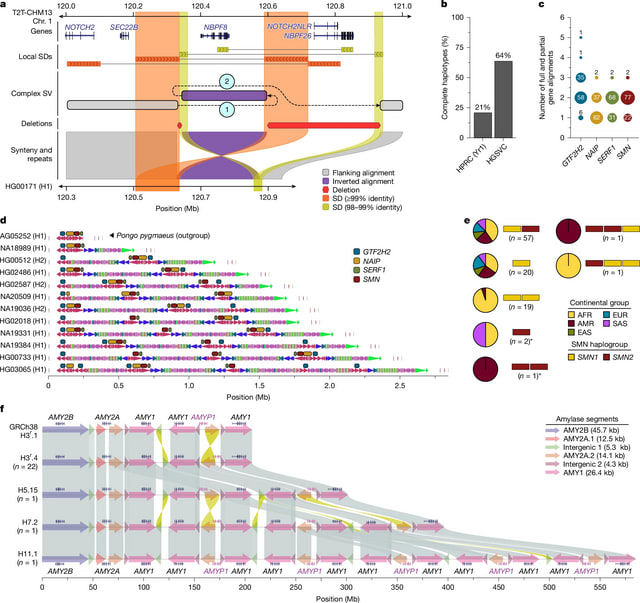

□ Complex genetic variation in nearly complete human genomes
>>
https://www.nature.com/articles/s41586-025-09140-6
They generated haplotype-resolved assemblies from all 65 diploid individuals using Verkko. The phasing signal was produced with Graphasing, leveraging Strand-seq to globally phase assembly graphs. The resulting haploid assemblies are highly contiguous at the base-pair level.
They integrated a range of quality control annotations for each assembly using established tools such as Flagger, NucFreq, Merqury and Inspector to compute robust error estimates for each assembled base.
To identify the centromeric regions within each Verkko and hifiasm (ultra-long) genome assembly, they first aligned the whole-genome assemblies to the T2T-CHM13 (v.2.0) reference genome using minimap2.
They built a pangenome graph of 214 haplotypes using Minigraph-Cactus (v.2.7.2) from haplotype-resolved assemblies of 65 HGSVC and 42 HPRC individuals, producing a CHM13-based VCF of top-level bubbles for genotyping with PanGenie.

□ The Human Organ Atlas
>>
https://www.biorxiv.org/content/10.1101/2025.07.31.667856v1
The Human Organ Atlas (HOA), an open data repository making accessible multiscale 3D imaging of human organs. The repository provides software tools and training resources enabling worldwide access, facilitating further research and the continued expansion of the HOA.
HOA employs a synchrotron imaging technique - Hierarchical Phase-Contrast Tomography (HiP-CT) that uses the ESRF's Extremely Brilliant Source, spanning whole organ imaging at around 20 um/voxel with local volumes of interest within the intact organs imaged down to ~ 1 um/voxel.

□ MViewEMA: Efficient Global Accuracy Estimation for Protein Complex Structural Models Using Multi-View Representation Learning
>>
https://www.biorxiv.org/content/10.1101/2025.07.25.666906v1
MViewEMA, a single-model EMA method that leverages a multi-view representation learning framework to integrate residue-residue interaction features from micro-environment, meso-environment, and macro-environment levels for global accuracy assessment of protein complex models.
MViewEMA operates without reliance on modeling-driven information sources. It employs specialized heterogeneous network architectures comprising graph, convolutional, and transformer modules to predict a global confidence score (i.e., TM-score) of the entire structure.

□ ProteinReasoner: A Multi-Modal Protein Language Model with Chain-of-Thought Reasoning for Efficient Protein Design
>>
https://www.biorxiv.org/content/10.1101/2025.07.21.665832v1
ProteinReasoner, a generative foundation model that incorporates structure and sequence as primary modalities, with the "evolutionary profile". ProteinReasoner integrates it as a central component of its reasoning process, analogous to chain-of-thought prompting in LLM.
ProteinReasoner captures the logic-driven tasks by modeling directional flows between modalities, including sequence → profile → structure and its reverse. It predicts the next structure token, the next amino acid, and the evolutionary profile of the subsequent position.

□ Taming the chaos gently: a predictive alignment learning rule in recurrent neural networks
>>
https://www.nature.com/articles/s41467-025-61309-9
“Predictive alignment” tames the chaotic recurrent dynamics to generate a variety of patterned activities via a biologically plausible plasticity rule.
Predictive alignment learning rule modifies plastic recurrent connections to predict output feedback signals, while aligning these predictive dynamics with existing chaotic spontaneous dynamics, which in turn suppress the chaos efficiently and improving network performance.
Predictive alignment trains networks to generate diverse complex target signals with nonlinear dynamics, such as the chaotic Lorenz attractor, delay-matching tasks that require short term memory of temporal information, and high-dimensional spatiotemporal patterns.

□ BioinAI: a general bioinformatic framework for multi-level transcriptomic data analysis using multiple semi-agents
>>
https://www.biorxiv.org/content/10.1101/2025.07.21.665890v1
BioinAl, a comprehensive bioinformatic framework comprising an online platform and two new algorithms, DeepAdvancer and stNiche. DeepAdvancer reconstructs the biologically meaningful gene expression profiles through weighted combinations of expression profiles from other classes.
Within DeepAdvancer, decoder weights are composed into a matrix whose dimensions correspond to the number of foundational classes multiplied by the number of genes.
This matrix serves as the central expression values for the foundational classes. A loss function is specifically included to minimize discrepancies between this generated matrix and the actual class-center values.
stNiche leverages spatial graph networks and symmetry-aware matching to identify spatial niches composed of diverse cell types, and further elucidates their functional roles and intercellular communication patterns.

□ DeepNanoHi-C: deep learning enables accurate single-cell nanopore long-read data analysis and 3D genome interpretation
>>
https://academic.oup.com/nar/article/53/13/gkaf640/8196083
DeepNanoHi-C, a novel deep learning framework specifically designed for scNanoHi-C data, which leverages a multistep autoencoder and a Sparse Gated Mixture of Experts (SGMoE) to accurately predict chromatin interactions by imputing sparse contact maps.
DeepNanoHi-C effectively captures complex global chromatin contact patterns through the multistep autoencoder and dynamically selects the most appropriate expert from a pool of experts based on distinct chromatin contact patterns.
DeepNanoHi-C integrates multiscale predictions through a dual-channel prediction net, refining complex interaction information and facilitating comprehensive downstream analyses of chromatin architecture.

□ TopoLa: A Universal Framework to Enhance Cell Representations for Single-cell and Spatial Omics through Topology-encoded Latent Hyperbolic Geometry
>>
https://www.biorxiv.org/content/10.1101/2025.07.23.666288v1
Topology-encoded Latent Hyperbolic Geometry (TopoLa), a novel framework designed to capture fine-grained intercellular relationships. Based on latent hyperbolic geometry, TopoLa models intercellular interactions in scRNA-seq and ST data through latent space embeddings.
The TopoLa framework demonstrates its transformative potential for assessing intercellular relationships. The topological similarities between cells (nodes) can be encoded into a latent hyperbolic space, enabling more precise measurement of the geometric structure of cell networks.
This conclusion is validated through proofs based on the principle of maximum entropy. Subsequently, the TopoLa distance (TLd) enables the determination of the positional distribution of cells in latent hyperbolic space.
TopoLa includes a component, spatial convolution via topology-encoded latent hyperbolic geometry (TopoConv), which utilizes TLd to convolve neighboring cells especially those with similar topological structures.
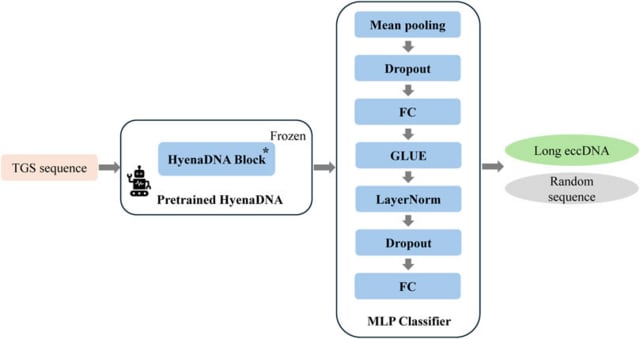

□ HyenaCircle: a HyenaDNA-based pretrained large language model for long eccDNA prediction
>>
https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1641162/full
HyenaCircle, a base-resolution prediction algorithm for long eccDNA formation, by adapting the HyenaDNA large language model architecture to third-generation sequencing data and full-length eccDNA sequences.
HyenaCircle achieved comparable performance with a validation AUROC of 0.715 and recall of 0.776. It surpassed DNABERT by 5.9% in AUROC and demonstrated stable convergence. Hyperparameter optimization confirmed batch size 16 and learning rate 5 × 10^−5 as optimal.

□ SimSpace: a comprehensive in-silico spatial omics data simulation framework
>>
https://www.biorxiv.org/content/10.1101/2025.07.18.665587v1
SimSpace, a flexible simulation framework that can generate synthetic spatial cell maps with categorical cell type labels and biologically meaningful organization.
Cell type spatial patterns are simulated using a Markov Random Field model, enabling the control of spatial autocorrelation and interaction between cell types. SimSpace captures a broad range of tissue architectures, from well-separated niches to spatially mixed environments.

□ Time-coexpress: temporal trajectory modeling of dynamic gene co-expression patterns using single-cell transcriptomics data
>>
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06218-w
TIME-CoExpress, a copula-based framework to model non-linear gene pair co-expression changes along cell pseudotime. A unique feature of this framework is its ability to accommodate covariate-dependent dynamic changes in correlation along cellular temporal trajectories.
TIME-CoExpress models dynamic gene zero-inflation patterns throughout cellular temporal trajectories. TIME-CoExpress captures the non-linear dependency between genes and to explore how predictor variables, such as cell pseudotime, influence gene-gene interactions.

□ DeepEVFI: Deep Evolutionary Fitness Inference for Variant Nomination from Directed Evolution
>>
https://www.biorxiv.org/content/10.1101/2025.07.22.666175v1
EVFI and Deep-EVFI infer variant fitness from time-series DNA sequencing data of variant frequencies using a temporal dynamics model, without relying on low-throughput, expensive functional measurements like binding affinity.
EVFI infers fitness using a masked optimization approach based on the presence of zero counts in consecutive timepoint pairs, which is equivalent to using conservative data-driven estimates.
DeepEVFI jointly learns a sequence-to-fitness neural network for fitness inference, using a conservative data-driven estimate, which they show improves inference for variants in the training set, evaluated on held-out selection rounds.

□ ScPGE: A scalable computational framework for predicting gene expression from candidate cis-regulatory elements
>>
https://www.biorxiv.org/content/10.1101/2025.07.21.666040v1
ScPGE (scalable computational framework for predicting gene expression from discrete candidate CREs) assembles DNA sequences, transcription factor (TF) binding scores, and epigenomic tracks from discrete cCREs into 3-dimensional tensors.
ScPGE models the relationships between CREs and genes by combining convolutional neural network with transformer. ScPGE directly puts chromatin loops into the self-attention layer, aiming to increase the attention weights of validated cCRE-gene interactions.
ScPGE uses an exponential decay function exp^-x/2 into chromatin loops, aiming to alleviate the sparsity of chromatin loops. A KL divergence loss between chromatin loops and attention weights is then added to the training loss, aiming to align their distributions.

□ MO-GCAN: Multi-Omics Integration based on Graph Convolutional and Attention Networks
>>
https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf405/8210085
MO-GCAN is an two-stage graph-based approach that integrates supervised feature learning followed by classification task by exploiting graph attention, convolutional network, and similarity network fusion.
After detecting the near-minimum threshold and a trained omics-specific model for each omics dataset, they forwarded the processed omics data and an affinity network to the chosen omics-specific GCN model to generate latent data for the selected omics.
MO-CCAN concatenates the latent data, constructed a fused similarity, detect a near-minimum threshold for the fused network to filter out weak connections, and put them to a graph attention network that employs two-head attention mechanism with the cross-entropy loss function.

□ DANCE 2.0: Transforming single-cell analysis from black box to transparent workflow
>>
https://www.biorxiv.org/content/10.1101/2025.07.17.665427v1
DANCE 2.0 addresses this urgent need by transforming single-cell preprocessing from a trial-and-error process into a systematic, data-driven, and interpretable workflow.
DANCE 2.0 consists of two core modules: the Method-Aware Preprocessing (MAP) module, which tailors preprocessing to specific downstream methods, and the Dataset-Aware Preprocessing (DAP), which recommends pipelines for new datasets via similarity-based matching.

□ Leviathan: A fast, memory-efficient, and scalable taxonomic and pathway profiler for next generation sequencing (pan)genome-resolved metagenomics and metatranscriptomics
>>
https://www.biorxiv.org/content/10.1101/2025.07.14.664802v1
Leviathan is a fast, memory-efficient, and scalable taxonomic and pathway profiler for next generation sequencing (genome-resolved) metagenomics and metatranscriptomics. Leviathanis powered by Salmon and Sylph in the backend.
Leviathan streamlines workflows for building taxonomic and functional profiling databases, profiling taxonomic and sequence abundance, profiling pathway abundance and coverage, and lazily merging sample-specific outputs into Xarray NeCDF and Apache Parquet artifacts.

□ Evaluation of sequencing reads at scale using rdeval
>>
https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf416/8210511
Rdeval can either run on the fly or store key sequence data metrics in tiny read 'snapshot' files. Statistics can then be efficiently recalled from snapshots for additional processing. Rdeval also generates a detailed visual report with multiple data analytics.
Rdeval can convert fa*[gz] files to and from other formats including BAM and CRAM for better compression. Overall, while CRAM achieves the best compression, the gain compared to BAM is marginal, and BAM achieves the best compromise between data compression and access speed.

□ cRegulon: Modeling combinatorial regulation from single-cell multi-omics provides regulatory units underpinning cell type landscape
>>
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03680-w
cRegulon infers regulatory modules by modeling combinatorial regulation of transcription factors based on diverse GRNs from single-cell multi-omics data.
cRegulon is introduced as a concept to integrate gene expression and epigenome state into regulatory units of gene regulation underlying cell types. It is formally defined as the TF combinatorial module as well as the RE that they bind to and the TGs that they regulate.

□ scSGC: Soft graph clustering for single-cell RNA sequencing data
>>
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06231-z
scSGC (Soft Graph Clustering for single-cell RNA sequencing data) aims to leverage soft graph construction to more accurately capture the continuous similarities between cells through non-binary edge weights.
scSGC facilitates improved identification of distinct cellular subtypes and clearer delineation of cell populations. scSGC utilizes a ZINB autoencoder to handle the sparsity and dropout issues inherent in scRNA-seq data, generating robust cellular representations.
Then, two soft graphs are constructed using the input data, and their corresponding laplacian matrices are computed. These matrices undergo a minimum jointly normalized cut through a graph-cut strategy to optimize the representation of cell-cell relationships.
scSGC employs an optimal transport-based self-supervised learning approach to refine the clustering, ensuring accurate partitioning of cell populations in high-dimensional and high-sparse data.

□ SubseqHash2: Efficient Seeding for Error-Prone Sequences
>>
https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf418/8211825
SubseqHash2, an improved algorithm that can compute multiple sets of seeds in one run, by defining k orders over all length-k subsequences and finding the optimal subsequence under each of the k orders in a single dynamic programming framework.
SubseqHash2 is further accelerated using SIMD instructions for parallel computing. The design of SubseqHash2 also allows it to generate the same sets of seeds for a string and its reverse complement by using symmetric random tables.
SubseqHash2 generates adequate seed matches for aligning hard reads, achieving high coverage of correct seeds and low coverage of incorrect seeds. Seeds produced by SubseqHash2 lead to more correct overlapping pairs at the same false-positive rate.

□ OmicsNavigator: an LLM-driven multi-agent system for autonomous zero-shot biological analysis in spatial omics
>>
https://www.biorxiv.org/content/10.1101/2025.07.21.665821v1
OmicsNavigator, an LLM-driven multi-agent system that autonomously distills expert-level biological insights from raw spatial omics data without domain-specific fine-tuning.
OmicsNavigator encodes spatial data into concise natural language summaries, enabling zero-shot annotation of structural components, quantitative analysis of pathological relevance, and semantic search of regions of interest using free-form text queries.

□ CellFuse Enables Multi-modal Integration of Single-cell and Spatial Proteomics Data
>>
https://www.biorxiv.org/content/10.1101/2025.07.23.665976v1
CellFuse, a deep learning-based, modality-agnostic integration framework designed specifically for settings with limited feature overlap.
CellFuse leverages supervised contrastive learning to learn a shared embedding space, enabling accurate cell type prediction and seamless integration across modalities and experimental conditions.

□ snATAC-Express infers Gene Expression from Prioritized Chromatin Accessibility Peaks using Machine Learning
>>
https://www.biorxiv.org/content/10.1101/2025.07.25.666784v1
snATAC-Express, a pipeline which trains machine learning models on snATAC-seq data to infer gene expression measured by snRNA-seq and to prioritize expression-relevant peaks.
The pipeline aggregates results from three machine learning approaches (random forest regression, XGBoost, and Light GBM) as well as linear regression to identify which ATAC peaks contribute to explaining variation among donors and cell types in pseudobulk gene expression.
Machine learning models outperform linear regression models, confirming that the relationship between chromatin accessibility and gene expression is more complex than simple correlation between increased accessibility and increased expression.

□ Parabricks: GPU Accelerated Universal Pan-Instrument Genomics Analysis Software Suite
>>
https://www.biorxiv.org/content/10.1101/2025.07.23.666378v1
Parabricks, a freely accessible, GPU-accelerated software suite supporting diverse workflows, including whole-genome, exome, transcriptome, and methylation analysis.
Parabricks is designed to streamline and accelerate a comprehensive range of genomic analysis modules by integrating industry-standard aligners such as BWA-MEM, Minimap2, and pangenome-aware Giraffe, as well as providing accelerated BWA-Meth for bisulfite sequencing.
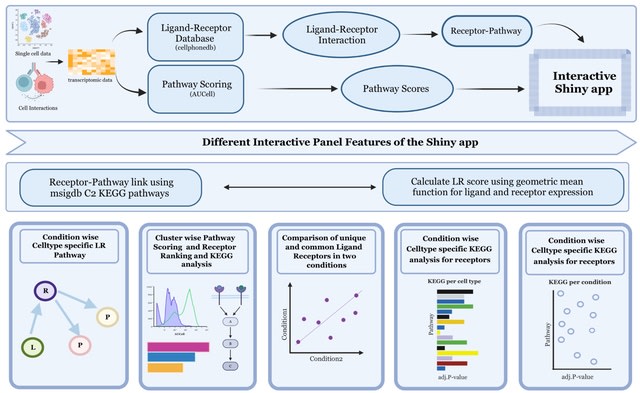

□ scVizComm: Pathway-Centric Visualization of Cell-Cell Communication in Single-Cell Transcriptomics Data
>>
https://www.biorxiv.org/content/10.1101/2025.07.25.666732v1
scVizComm, an interactive visualization tool to display pathway and associated ligand-receptor interactions. scViZComm visualises condition-wise Ligand-Receptor interaction for the source and target clusters of choice, and determines expression dependent LR Score.
scVizComm features distribution of genes associated with the selected pathway using AUCell, and KEGG pathway analysis for the receptors associated per cluster or condition, thereby deter-mining the downstream of the receptor.

□ CYCLONE: recycle contrastive learning for integrating single-cell gene expression data
>>
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06214-0
CYCLONE, a new method for integrating single-cell gene expression data using a recycle contrastive learning network. The contrastive learning network and the VAE model work together to jointly train the low-dimensional representations.
CYCLONE iteratively updates the network parameters using gradient backpropagation to navigate the low-dimensional space, gradually reducing noise. This recycle update process enhances the accuracy of positive sample pairs, effectively guiding batch effect removal.
CYCLONE constructs positive sample pairs by augmenting MNN pairs with KNN pairs identified within batches, thereby expanding the range of covered cell types.






































































































