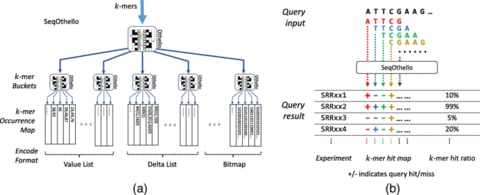
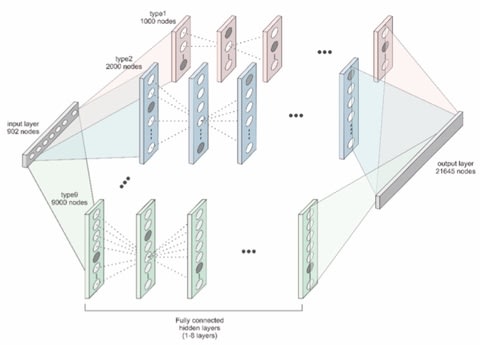
全ての事象は散逸構造の中の計算過程として観察される。だが多体間の相関性は、一元構造の内部に掘り出されるシミュラークルに等しく、時間とは可変な測度である以上の意味を為さない。

□ Rev D: Oxford Nanopore has released a new version of MinION and GridION flow cells that include the new ‘Rev D’ ASIC.
>>
https://nanoporetech.com/about-us/news/oxford-nanopore-releases-rev-d-flow-cells-enabling-increase-data-yields
Rev D extends the amount of time that flow cells can be used for DNA sequencing or RNA sequencing, increasing the overall yields of DNA sequence data to as much as 30 Gb per flow cell (at this performance, the equivalent of ~10X human genome for $500*). Rev D increases the rate at which DNA fragments pass through a nanopore from 35 bases per second at launch to 450 bases per second now.
□ Do Cells use Passwords? Do they Encrypt Information?:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/432120.full.pdf
Encryption could benefit cells by making it more difficult for pathogens to hijack cell networks. Because the 'language' of cell signaling is unknown, i.e., similar to an alien language detected by SETI. use information theory to consider the general case of how non-randomness filters can be used to recognize (1) that a data stream encodes a language, rather than noise, and (2) quantitative criteria for whether an unknown language is encrypted.
This leads to the result that an unknown language is encrypted if efforts at decryption produce sharp decreases in entropy and increases in mutual information. The magnitude of which should scale with language complexity. demonstrate this with a simple numerical experiment on English language text encrypted with a basic polyalphabetic cipher.
□ Caring without sharing: Meta-analysis 2.0 for massive genome-wide association studies:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/05/436766.full.pdf
The extension to this methods and paradigm would be to adopt an honest-but-curious threat model for all parties and aim to prevent leakage of individual level information from the data owning silos to any other party involved. Since for all steps of the pipeline the central hub simply sums the results of all silos (or takes averages), when more than 2 silos contribute, multi-party, secure sum protocols can be used to decrease the overall chance of information leakage.
□ Improved DNA based storage capacity and fidelity using composite DNA letters:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/02/433524.full.pdf
The Shannon information capacity of DNA was recently demonstrated, using fountain codes, to be ~1.57 bit per synthesized position. However, synthesis and sequencing technologies process multiple nominally identical molecules in parallel, leading to significant information redundancies. composite DNA alphabets, using mixed DNA base types, to leverage this redundancy, enabling higher density. develop encoding and decoding for composite DNA based storage, including error correction. Using current DNA synthesis technologies, they code 6.4 Megabyte data into composite DNA, achieving ~25% increase in capacity as compared to literature.

□ MiniScrub: de novo long read scrubbing using approximate alignment and deep learning:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/02/433573.full.pdf
a novel Convolutional Neural Network (CNN) based method, called MiniScrub, for de novo identification and subsequent “scrubbing” (removal) of low-quality Nanopore read segments. MiniScrub first generates read-to-read alignments by MiniMap, then encodes the alignments into images, and finally builds CNN models to predict low-quality segments that could be scrubbed based on a customized quality cutoff. Compared to raw reads, de novo genome assembly with scrubbed reads pro- duces many fewer mis-assemblies and large indel errors.

□ poreTally: run and publish de novo Nanopore assembler benchmarks:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/09/23/424184.full.pdf
poreTally automatically assembles a given read set using several often-used assembly pipelines, analyzes the resulting assemblies for correctness and continuity, and finally generates a quality report. For the running of assembly pipelines, poreTally relies on the Snakemake work-flow management system and its excellent integration with conda environments.
□ Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/434118.full.pdf
determined the precision and recall, present high confidence and high sensitivity call sets of variants and discuss optimal parameters. The aligner Minimap2 and structural variant caller Sniffles are both the most accurate and the most computationally efficient tools. While LAST is the recommended aligner by the authors of NanoSV the number of variants identified was excessive, with a high number of false positives. NanoSV, too, obtained the best results after minimap2 alignment.
In this comparison with SVs called from short read sequencing data using Manta and Lumpy a clear advantage for long reads was demonstrated, with substantially higher recall values.
□ rCASC: reproducible Classification Analysis of Single Cell sequencing data:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/430967.full.pdf
rCASC a modular RNAseq analysis workflow allowing data analysis from counts generation to cell sub-population signatures identification, granting both functional and computation reproducibility. CASC uses as core application to detect cell clusters the “kernel based similarity learning”: identification of the optimal number of clusters for cell partitioning. The evaluation of clusters stability, measuring the permanence of a cell in a cluster upon random removal of subsets of cells.
□ Time and space dimensions of gene dosage imbalance of aneuploidies revealed by single cell transcriptomes:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/09/23/424887.full.pdf
gene dosage imbalance is of bidimensional nature: over time (simultaneous expression of all alleles resulting in increased accumulation of RNA of copy altered genes) as previously stated, and over space (increased fraction of cells simultaneously expressing copy altered genes).

□ fastp: an ultra-fast all-in-one FASTQ preprocessor:
>>
https://academic.oup.com/bioinformatics/article/34/17/i884/5093234
fastp provides functions including quality profiling, adapter trimming, read filtering and base correction, and supports both single-end and paired-end short read data and also provides basic support for long-read data. fastp includes most features of FASTQC + Cutadapt + Trimmomatic + AfterQC while running 2–5 times faster than any of them alone.
□ SemGen: a tool for semantics-based annotation and composition of biosimulation models:
>>
https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/bty829/5107020
A key SemGen capability is to decompose and then integrate models across existing model exchange formats including SBML and CellML. To support this capability, using semantic annotations to explicitly capture the underlying biological and physical meanings of the entities and processes that are modeled. SemGen leverages annotations to expose a model’s biological and computational architecture and to help automate model composition.
□ Grouper: graph-based clustering and annotation for improved de novo transcriptome analysis:
There are two main modules in Grouper: the clustering module and the labeling module. The former is based on the tool, RapClust, and is designed to be run downstream of the Sailfish or Salmon tools for rapid transcript-level quantification. It relies on the fragment equivalence classes, orphaned read mappings and quantification information computed by these tools in order to determine how contigs in the assembly are potentially related and cluster them accordingly.


□ Energy performance optimization in buildings: A review on semantic interoperability, fault detection, and predictive control:
>>
https://aip.scitation.org/doi/full/10.1063/1.5053110
The traditional architecture of BAS is often represented as a three-layer architecture: the field layer includes sensors, actuators, and controllers interconnected via field buses like KNX, LON, or wireless networks like ZigBee or Z-Wave. The automation layer consists of PLCs covering measurement processing, control, and alarm tasks for the devices of the field layer and uses protocols of both the field and the management layer. The management layer forms the upper tier of the architecture and is constituted of supervisory control systems (SCS), human-machine interfaces (HMI) with configuration and monitoring features, as well as databases for time series data archival (DBs). Typical protocols of the management layer are BACnet or OPC.
□ rMETL: sensitive mobile element insertion de- tection with long read realignment:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/09/19/421560.full.pdf
rMETL takes advantage of its novel chimeric read re-alignment approach to well handle complex MEI signals. Benchmarking results demonstrate that rMETL can produce high quality callsets to improve long read-based MEI calling.

□ scClustViz: Single-cell RNAseq cluster assessment and visualization:
>>
https://f1000research.com/articles/7-1522/v1
Given that cell types are expected to have distinct gene expression patterns, scClustViz uses differential gene expression between clusters as a metric for assessing the fit of a clustering result to the data at multiple cluster resolution levels. scClustViz provides interactive visualisation of cluster-specific distributions of technical factors, predicted cell cycle stage and other metadata; cluster-wise gene expression statistics to simplify annotation and identification specific marker genes; and GE distributions.

□ Genomic prediction of autotetraploids; influence of relationship matrices, allele dosage, and continuous genotyping calls in phenotype prediction:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/432179.full.pdf
Continuous genotypic based models performed as well as the current best models and presented a significantly better goodness-of-fit for all traits analyzed. This approach also reduces the computational time required for marker calling and avoids problems associated with misclassification of genotypic classes when assigning dosage in polyploid species.

□ scRNA-seq mixology: towards better benchmarking of single cell RNA-seq protocols and analysis methods:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/433102.full.pdf
a realistic benchmark experiment that included mixtures of single cells or ‘pseudo-cells’ created by sampling admixtures of cells or RNA from 3 distinct cancer cell lines. The comparison shows the 10X Chromium platform to produce the highest quality data, both Drop-seq and CEL-seq2 are very flexible protocols, with various parameters that can be optimized and tuned.
systematic methods comparisons for 4 key tasks; normalization and imputation, clustering, trajectory analysis and data integration. The performance of methods varied across different datasets, with no clear winners in all situations, however, consistently satisfactory results were observed for scran, Linnorm, DrImpute and SAVER for normalization and imputation; Seurat and SC3 for clustering; Monocle2 and Slingshot for trajectory analysis and MNN for data integration.
□ mirtronDB: a mirtron knowledge base:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/429522.full.pdf
Mirtrons are originated from short introns with atypical cleavage from the miRNA canonical pathway by using the splicing mechanism. the first knowledge database dedicated to mirtron, called mirtronDB, has a total of 1,407 mirtron precursors and 2,426 mirtron mature sequences in 18 species.
□ dream: Powerful differential expression analysis for repeated measures designs:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/432567.full.pdf
The dream model extends
• multiple random effects
• the variance terms to vary across genes
• estimate residual degrees of freedom for each model from the data in order to reduce false positives
• hypothesis testing with moderated t-statistics using empirical Bayes approach
• fast hypothesis testing for fixed effects in linear mixed models
• small sample size hypothesis test to increase power
• precision weights to model measurement error in RNA-seq counts
• seamless integration with the widely used workflow of limma
□ Novel Data Transformations for RNA-seq Data Analysis:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/432690.full.pdf
Simulation studies showed that limma based on transformed data by using the rv transformation performed best compared with limma based on transformed data by using other transformation methods in term of high accuracy and low FNR, while keeping FDR at the nominal level. For large sample size, limma with the r2 transformation performed better than limma with the voom transformation. In real data analysis, several (l2, l, r2, r, rv, and rv2) of our proposed transformations performed better than voom.

□ Bazam : A rapid method for read extraction and realignment of high throughput sequencing data:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/03/433003.full.pdf
Bazam, a tool that efficiently extracts the original paired FASTQ from reads stored in aligned form (BAM or CRAM format). Bazam extracts reads in a format that directly allows realignment with popular aligners with high concurrency. Bazam increases parallelism by splitting the output streams into multiple paths for separate realignment. a single source alignment can be realigned using an unlimited number of parallel aligners, significantly accelerating the process when a computational cluster is available. Through eliminating steps and increasing the accessible concurrency, Bazam facilitates up to a 90% reduction in the time required for realignment.

□ GPyTorch beta: Scalable Gaussian processes in PyTorch, with strong GPU acceleration.
>>
https://gpytorch.ai/
GPyTorch provides significant GPU acceleration (through MVM based inference); implementations of the latest algorithmic advances for scalability and flexibility (SKI/KISS-GP, stochastic Lanczos expansions, LOVE, SKIP, stochastic variational deep kernel learning, ...)

□ An information theoretic treatment of sequence-to-expression modeling:
>>
https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006459
Methodologically, an important feature of this approach was the generation of ensemble by uniform sampling in the multi-dimensional space, followed by optimization, as was done in. Using the fact that the entropy of the probability distribution captures the uncertainty intrinsic, we can use the difference in entropy of the original ensemble & the filtered ensemble (information gain), as an objective evaluation of how informative the experiments results are.
□ Improving on hash-based probabilistic sequence classification using multiple spaced seeds and multi-index Bloom filters:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/05/434795.full.pdf
a novel probabilistic data structure based on Bloom filters that implicitly stores hashed data (to reduce memory usage) yet can better handle sequence polymorphisms and errors with multiple spaced seeds, increasing the sensitivity of hashed-based sequence classification. multi-index Bloom Filter shows a higher sensitivity and specificity for read- binning than BWA MEM at an order of magnitude less time. For taxonomic classification, miBF shows higher sensitivity than CLARK-S at an order of magnitude less time while using half the memory.
□ Quasi-universality in single-cell sequencing data:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/10/05/426239.full.pdf
This direct approach across technologies, roughly 95% of the eigenvalues derived from each single-cell data set can be described by universal distributions predicted by Random Matrix Theory. Interestingly, 5% of the spectrum shows deviations from these distributions and present a phenomenon known as eigenvector localization, where information tightly concentrates in groups of cells.