










(A Hybrid Solar Eclipse over Kenya
Image Credit & Copyright: Eugen Kamenew)
□ A Hybrid Solar Eclipse over Kenya
>> https://apod.nasa.gov/apod/ap170724.html
the Moon covers much of the Sun during a hybrid eclipse, a rare type of solar eclipse that appears as total from some Earth locations, but annular in others. During the annular part of the eclipse, the Moon was too far from the Earth to block the entire Sun. Next month a total solar eclipse will cross the USA.

(iPhone camera.)
dawn begins to creep.

□ DeepSF: deep convolutional neural network for mapping protein sequences to folds:
>> https://arxiv.org/pdf/1706.01010.pdf
a 1-dimensional deep convolution neural network to classify proteins of variable-length into all 1195 known folds defined in SCOP 1.75 DB. Each residue in a sequence is represented as a 20-dimension zero-one vector, in which only the value at the residue index is marked as 1 and all others are marked as 0.

□ Consistency of heterogeneous synchronization patterns in complex weighted networks:
>> http://aip.scitation.org/doi/figure/10.1063/1.4977972
The coexistence of synchronizations is a phenomenon in which a variety of complex functional relationships are established, between the dynamical evolutions of some coupled oscillatory elements.
□ Deep Learning: Generalization Requires Deep Compositional Feature Space Design:
>> https://arxiv.org/pdf/1706.01983.pdf
Increasing the number of convolution kernel for a layer doesn’t necessarily solve the problem. Before reducing the feature space size it’s important to project into high dimensional hyperspaces using multiple convolution operations.
□ PathCORE: Visualizing globally co-occurring pathways in large transcriptomic compendia:
>> http://biorxiv.org/content/biorxiv/early/2017/06/08/147645.full.pdf
PathCORE is a hypothesis generation tool that identifies co-occurring pathways from the results of unsupervised analysis. In PathCORE, pathway annotations undergo a procedure called maximum impact estimation.
□ Indexcov: fast coverage quality control for whole-genome sequencing
>> http://biorxiv.org/cgi/content/short/148296v1
Indexcov provides facilitates the detection of coverage anomalies such as aneuploidies, megabase-scale deletions and duplications. Absolutely--that's actually the reason for it! Big innovations (all credit to @brent_p) are using index (<1s speed) & viz/quantification

□ PhenoSpD: an atlas of phenotypic correlations and a multiple testing correction for the human phenome:
>> http://biorxiv.org/content/biorxiv/early/2017/06/10/148627.full.pdf
PhenoSpD multiple testing correction function provides a conservative way to reduce dimensionality for GWAS of complex molecular traits. PhenoSpD builds on the phenotypic correlation function of metaCCA and LD score regression using only GWAS summary results. It can also be used as an indicative threshold for post-GWAS data mining tools such as MR-Base and LD Hub.

□ Gene annotation bias impedes biomedical research
>> http://biorxiv.org/content/early/2017/05/02/133108
Striking correlation of published disease-gene associations with annotations, not with gene expression effect size. this experimental paradigm has led to a gene-centric disease research bias where hypotheses are confounded by the “streetlight effect”. “answers where the light is better rather than where the truth is more likely to lie”.
To enable researchers to pursue data-driven hypotheses, they have made GE meta-analysis data publicly available at MetaSignature.
□ DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data:
>> http://biorxiv.org/content/biorxiv/early/2017/06/12/149328.full.pdf
For the short sequence reads model, the input of the DIAMOND alignment consists of the 100nt long khmers extracted from the UNIPORT genes. the alignment stage only passes ARG-like sequences that have e-value<1e-10 and identity>20% to deepARG for prediction.
□ Parallel Computing to Speed up Whole-Genome Bayesian Regression Analyses Using Orthogonal Data Augmentation:
>> http://biorxiv.org/content/biorxiv/early/2017/06/12/148965.full.pdf
a strategy to parallelize Gibbs sampling for each marker within each step of the MCMC chain.
□ Non-Parametric Genetic Prediction of Complex Traits with Latent Dirichlet Process Regression Models:
>> http://biorxiv.org/content/biorxiv/early/2017/06/13/149609.full.pdf
despite its faster computational speed, the VB algorithm is less accurate than MCMC when SNP heritability is large, sometimes by quite a large margin (e.g. PVE=0.80 in simulations). Posterior independence is an unrealistic assumption given that SNP genotypes are correlated through LD.
□ Revisiting the General Concept of Network Centralities: A Propose for Centrality Analysis in Network Science:
>> http://biorxiv.org/content/early/2017/06/13/149492.full.pdf
the determination of important nodes within a network depends on its topology. Centralities of Lobby index, ClusterRank, Laplacian, MNC, Degree, Markov, Diffusion degree, Kleinberg's hub, Eigen vector, Authority score, Katz centralities fell in the same group where all the silhouette scores are more than the average equal to 0.61.
□ vqtl: An R package for Mean-Variance QTL Mapping:
>> http://biorxiv.org/content/biorxiv/early/2017/06/13/149377.full.pdf
The permutation function, scanonevar.perm, executes a carefully-constructed set of permutations to empirically estimate a FWER-controlling p-value for each observed LOD score.
□ LIONS: Analysis Suite for Detecting and Quantifying Transposable Element Initiated Transcription from RNA-seq:
>> http://biorxiv.org/content/biorxiv/early/2017/06/13/149864.full.pdf
Further refinement of these methods such as inclusion of aligned-strand bias measures, position-aware Hidden Markov Model or machine-learning trained sorting algorithms to detect the molecular signature of TSSs could be used to improve the transcript assembly.
□ Fractional Langevin Monte Carlo: Exploring Lévy-driven Stochastic Differential Equations for Markov Chain Monte Carlo
>> https://arxiv.org/pdf/1706.03649v1.pdf
FLMC framework is based on a family of heavy-tailed distributions, called α-stable Lévy distributions. FLMC can provide superior performance in multi-modal settings, improved convergence rates, and robustness to algorithm parameters.
□ K-mer clustering algorithm using a MapReduce framework: the parallelization of the Inchworm module of Trinity
>> http://biorxiv.org/content/early/2017/06/15/149948
□ Modeling of cytometry data in logarithmic space: when is a bimodal distribution not bimodal?:
>> http://biorxiv.org/content/biorxiv/early/2017/06/14/150201.full.pdf
automatically cluster or build dynamical models which rely on an apparent bi-modal structure, as it might depend on whether the data was log-transformed or not. increasingly relevant as cytometry moves forward to higher dimensional measurements which become tractable only w/ automatic gating schemes.

□ jkpritch:
In our omnigenic model we argue that most of the genome is near to genes that affect any given trait (1/n)
>> http://www.cell.com/cell/abstract/S0092-8674(17)30629-3
□ 16GT: new super-fast and sensitive variant caller using a 16-genotype probabilistic model in a single algorithm:
>> https://academic.oup.com/gigascience/article-lookup/doi/10.1093/gigascience/gix045
For indels, 16GT produced slightly fewer true positive calls and slightly more false negative calls than HaplotypeCaller, but less than half as many false negative calls as UnifiedGenotyper.

□ Out-of-equilibrium statistical dynamics of spatial pattern generating cellular automata:
>> http://biorxiv.org/content/biorxiv/early/2017/06/16/151050.full.pdf
Lattices of cells behave as particles stochastically rolling down a pseudo-energy landscape – defined by a spin glass-like Hamiltonian. A new kinetic mechanism “pathway trapping” yields metastable spatial patterns by preventing minimization of the particle’s pseudo-energy.

□ Scalable composition frameworks for multicellular:
>> http://biorxiv.org/content/biorxiv/early/2017/06/16/150987.full.pdf
CALIN is a tool for automated design of DNA architectures allowing the implementation of multicellular N-inputs logic functions. the systematic multicellular consortia performs recombinase-based Boolean / history-dependent logic, and integrating an arbitrary N-inputs.

□ Addressing the looming identity crisis in single cell RNA-seq:
>> http://www.biorxiv.org/content/biorxiv/early/2017/06/16/150524.full.pdf
MetaNeighbor allows to quantify the degree to which cell types replicate across datasets, and rapidly identify clusters w/ high similarity. their AUROC scores are exceptionally high, particularly when considered in the context of the well-described technical confounds of sc-data. They suspect this reflects the fundamental nature of the biological problem we are facing, discrete cell types can be identified by the transcriptional profiles / the biological clarity of the problem overcomes technical variation.
□ EPSILON-CP: using deep learning to combine information from multiple sources for protein contact prediction:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1713-x
a meta prediction method EPSILON-CP combines physicochemical, evolutionary & sequence-based information, is extending over current methods. EPSILON-CP achieves a mean precision of 0.305 on L/5, compared to 0.284 for MetaPSICOV. EPSILON-CP outperforms the co-evolutionary methods 5-fold with an accuracy of 53.1% on L/5 compared to 8.3% for GaussDCA.
□ FactorNet: a deep learning for predicting cell type specific TF binding from nucleotide-resolution sequential data:
>> http://biorxiv.org/content/biorxiv/early/2017/06/18/151274.full.pdf
the directional nature & modularity of DNA sequences prompt to search for a model that can discover local patterns & long-range interactions, which led us to ultimately select a hybrid neural network architecture that includes convolution and bidirectional recurrence.
□ Neural Network and Nearest Neighbor Algorithms for Enhancing Sampling of Molecular Dynamics:
>> http://pubs.acs.org/doi/abs/10.1021/acs.jctc.7b00188
this method is capable of achieving ergodic sampling and calculating free energy of polypeptides with up to 8-dimensional bias potential.
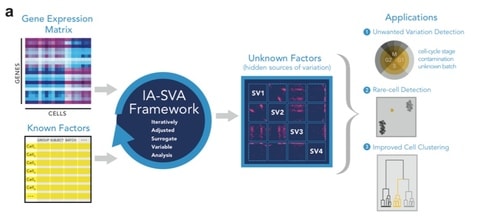
□ IA-SVA: A robust statistical framework to detect multiple sources of hidden variation in single-cell transcriptomes:
>> http://biorxiv.org/content/biorxiv/early/2017/06/18/151217.full.pdf
□ One Model To Learn Them All: building multi-domain systems
>> https://arxiv.org/abs/1706.05137

□ Fine-mapping identifies causal variants for RA and T1D in DNASE1L3, SIRPG, MEG3, TNFAIP3 and CD28/CTLA4 loci:
>> http://biorxiv.org/content/biorxiv/early/2017/06/19/151423.full.pdf
annotations in narrowPeak format for H3K4me3 peaks, DNAse-I peaks, & ChromHMM genome segmentations from the Roadmap epigenetics consortium. For 50% (=10/20) of RA and 72% (=26/36) of T1D loci, the most significant variant was in linkage disequilibrium (LD; r2>0.8).

□ HetNet: Integrating All of Biology into a Public Neo4j Database:
>> https://neo4j.com/blog/integrating-biology-public-neo4j-database/
HetNet has 50,000 nodes of 11 types which we would call labels in Neo4j. Btwn these 50,000 nodes are 2.25 million relationships of 24 types. The way they decided to understand the relationship btwn a compound & a disease is to look along certain types of metapaths.
□ Implementation of nanonet using keras.
>> https://gist.github.com/cjw85/56f6aae3d3ce8995af6f8173b6f4eb07
Reuses bits of nanonet for peripheral calculations and decoding. Overall speed is limited by decoding step. Reads are chunked into a maximum of 1000 feature vectors for processing on GPU.
□ An open-source protein database that you can start using with Keras models in 2 lines of code:
>> https://github.com/PeptoneInc/dspp-keras
Protein order and disorder data for Keras and Tensor Flow frameworks with automated update cycle made for continuous learning applications.
□ Rosetta:MSF: a modular framework for multi-state computational protein design:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005600
□ GfaPy: a flexible and extensible software library for handling sequence GFA (Graphical Fragment Assembly) graphs in Python:
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btx398/3883940/GfaPy-a-flexible-and-extensible-software-library
GfaPy is a software package for creating, parsing and editing Graphical Fragment Assembly graphs using the programming language Python.
def linear_paths(self, redundant_junctions=False):
"""Find linear paths of dovetail overlaps connecting segments.
redundant_junctions (bool): output the junction segments at the end of each path which involves them; this mimics the construction of contigs in string graph assemblers Readjoiner and SGA; default: False"""
□ A Bayesian Framework for Multiple Trait Colocalization from Summary Association Statistics
>> http://biorxiv.org/content/early/2017/06/26/155481
□ Combining Semantic Similarity and GO Enrichment for Computation of Functional Similarity:
>> http://biorxiv.org/content/biorxiv/early/2017/06/26/155689.full.pdf
GO enrichment assumes a hypergeometric distribution of annotating terms in a gene set & has been effectively used in finding pathways. the local context of querying genes is sensitive to the missing and spurious terms in the GO annotating corpus.
□ Deep Semantic Role Labeling: a deep highway BiLSTM architecture with constrained decoding.
>> https://homes.cs.washington.edu/~luheng/files/acl2017_hllz.pdf
the 8-layer BiLSTMs incorporated some of the recent best practices such as orthonormal initialization, RNN-dropout, and highway connections. consists of 4 forward LSTMs & 4 reversed LSTMs w/ 300-dimensional hidden units, and a softmax layer for predicting the output distribution.
□ MolecuLeNet: a novel deep learning architecture modeling quantum interactions in molecules
>> https://arxiv.org/abs/1706.08566
the continuous-filter convolutional layers in the architecture respects quantum-chemical constraints, such as rotationally invariant energy predictions as well as rotationally equivariant, energy-conserving force predictions.

□ MassBlast: A workflow to accelerate RNA-seq and DNA database analysis:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/03/131953.full.pdf
MassBlast requires BLAST+ and a Ruby environment with bioinformatic libraries, Bioruby and ORF Finder. It can filter the results and significantly improve the annotation of multiple sequencing databases for exploring new biosynthetic pathways.

□ Toward A Scalable Exploratory Framework for Complex High-Dimensional Phenomics Data:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/05/159954.full.pdf
it uses emerging principles from algebraic topology as the basis to observe and discern structural features from high-dimensional data. notably, this approach shows how the environment plays a key role in determining the phenotypic behavior of one of the two genotypes.

□ Intervene: a tool for intersection and visualization of multiple gene or genomic region sets:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1708-7
Intervene’s pairwise module provides several metrics to assess intersections, including number of overlaps, fraction of overlap, Jaccard statistics, Fisher’s exact test, and distribution of relative distances.