“The survival ... of certain favored words in the struggle for existence is natural selection,”
- Charles Robert Darwin.
□ Ages and cycles of nature in ceaseless sequence moving. Ceiling detail at National Academy of Sciences
□ Low frequency fully kinetic simulation of the toroidal ion temperature gradient instability
>>
http://ow.ly/4Rpv30fVYMO
□ A Bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data:
>>
https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btx605/4222631/A-Bayesian-framework-for-the-inference-of-gene?redirectedFrom=fulltext
a first-order autoregressive moving-average model and it infers the gene regulatory network within a variational Bayesian framework.
GRNVBEM/HYPERPARAMETERS
gene expression time-series data. Gx(N+1) real matrix with the fold change of G genes measured at (N+1) time points.
□ Using Seurat with multi-modal data:
>>
http://satijalab.org/seurat/multimodal_vignette.html
For CITE-seq data, a centered log-ratio (CLR) normalization computed independently for each gene.
cbmc<-NormalizeData(cbmc, assay.type="CITE",normalization.method="genesCLR")
cbmc<-ScaleData(cbmc, assay.type="CITE",display.progress=FALSE)
□ 10x Genomics and the Human Cell Atlas International Consortium Announce Partnership:
>>
https://www.10xgenomics.com/news/10x-genomics-human-cell-atlas-international-consortium-announce-partnership/
□ GRAF, a new tool for finding duplicates and closely related samples in large genomic datasets
>>
https://ncbiinsights.ncbi.nlm.nih.gov/2017/10/16/graf-new-tool-finds-duplicates-close-relatives-large-genomic-datasets-dbgap/
GRAF can be used to validate the relationships of subjects and samples reported in the pedigree file and the subject-sample mapping file (SSM) against the genotype data.
□ Illumina Ventures Raises $230M for First Fund:
>>
https://www.genomeweb.com/sequencing/illumina-ventures-raises-230m-first-fund
□ Nanopack: a great softwarepack for the analysis of your Nanopore data
>>
http://github.com/nanopack

□ The latest verison of the MaSuRCA assembler supports nanopore reads
>>
http://bit.ly/2ySTwgg
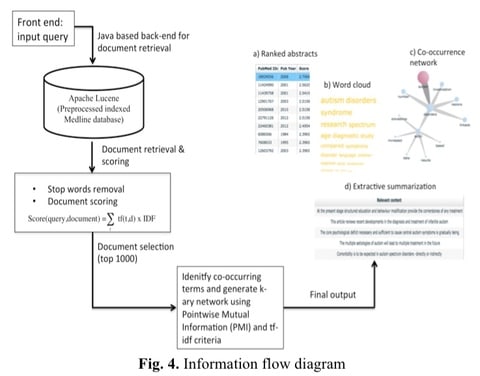
□ SEACOIN 2.0: interactive mining / visualization tool for information retrieval, summarization & knowledge discovery:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/19/206193.full.pdf
SEACOIN 2.0 has the potential to generate novel hypothesis and discover previously unknown connections btwn diseases, genes, chemicals, etc. A multi-level interactive topological visualization is used to present the k-ary relation network where each node represents biomedical/clinical terms associated with the query. BioCreative IV corpus can accurately retrieve most relevant documents related to the query without generating too many false positives.
□ CSER: Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine:
>>
http://www.cell.com/ajhg/fulltext/S0002-9297(16)30106-9
□ Two distinct DNA sequences recognized by transcription factors represent enthalpy and entropy optima:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/19/205906.full.pdf
The common presence of at least two local optima is general to all macromolecular interactions, as ΔG depends on two partially independent variables ΔH and ΔS according to the central equation of thermodynamics, ΔG = -RTlnKd = ΔH - TΔS.

□ Darwin Assembly: fast, efficient, multi-site bespoke mutagenesis:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/21/207191.full.pdf
using a non-mutagenic “θ” boundary, the library was amplified, cloned and transformed with an estimated 2.25x10^8 transformants generated. Darwin Assembly is amenable to automation, as most steps are enzymatic in compatible buffers and can readily be programmed.
□ Effects of demographic stochasticity and life-history strategies on times and probabilities to fixation:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/21/207100.full.pdf
For both of these definitions, fitness is a quantity that is not inherent to the individual but depends on 1-side on demographic parameters and on the other side on both the population size and, in a non neutral framework, its genetic composition.
□ DeepMind AI has mastered 3,000 years of human knowledge in 40 hours. Next up is drug discovery
>>
https://bloom.bg/2yGEcCB

□ Evidence of reduced recombination rate in human regulatory domains
>>
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1308-x
the existence of a recombination rate valley at regulatory domains, consisting of regulatory elements and their target genes. and provide a potential molecular mechanism to interpret the interplay between genetic and epigenetic variations.

□ RACIPE: A computational tool for Modeling Gene Regulatory Circuits using Randomization:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/27/210419.full.pdf
RACIPE integrates statistical learning methods with parameter perturbations, which makes it distinct from the traditional parameter sensitivity analysis, parameter space estimation and other randomization strategies.
□ FAST-SG: An alignment-free algorithm for hybrid assembly:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/26/209122.full.pdf
The core of FAST-SG is an alignment-free algorithm specifically designed to construct the scaffolding graph from either short or long reads using light-weight data structures.
□ Toyota Signs Licensing Agreement with Kazusa DNA Research Inst, Eurofins Genomics & GeneBay for GRAS-Di DNA Analysis
>>
http://www.asiaone.com/business/toyota-signs-licensing-agreement-kazusa-dna-research-institute-eurofins-genomics-and
GRAS-Di: Genotyping by Random Amplicon Sequencing-Direct.
A device capable of analyzing tens of millions to billions of locations on the genome at the same time.
□ トヨタ自動車 かずさDNA研究所、ユーロフィンジェノミクス、ジーンベイとDNA解析技術「GRAS-Di」のライセンス契約を締結
>>
http://newsroom.toyota.co.jp/jp/detail/19434333
□ Active deep learning reduces annotation burden in automatic cell segmentation:
>>
https://www.biorxiv.org/content/early/2017/10/29/211060
In uncertainty sampling, the learner queries the user or annotator to label the most informative samples. The algorithm queries the instance whose prediction is the least confident:
x*LC = argmaxx(1-Pθ (y|x))
where, y = argmaxyPθ(y|x) , or the class label with the highest posterior probability under the model θ.
□ runibic: a Bioconductor package for parallel row-based biclustering of gene expression data:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/28/210682.full.pdf
The proposed method based on a stable STL sort, could be used as an alternative to the C style pointer and sorting based on Fibonacci Heap.
library(runibic)
library(biclust)
test<−matrix(rnorm(1000), 100, 100)
res<−runibic(test)
res<−biclust : : biclust(test, method = BCUnibic())
□ BGI's MGI Tech Launches Two New NGS Platforms:
>>
http://markets.businessinsider.com/news/stocks/BGI-s-MGI-Tech-Launches-Two-New-NGS-Platforms-1006267619
□ LULU: Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates
>>
https://www.nature.com/articles/s41467-017-01312-x
the aim to retain true α-diversity and taxonomic composition, while discarding the artefactual OTUs from community data derived by HTS of marker genes (metabarcoding).
□ Targeting the Untargetable: Predicting Pramlintide Resistance Using a Neural Network Based Cellular Automata:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/30/211383.full.pdf
The trained model is implanted into cells in a 2-dimensional on-lattice agent based model and is used to model phenotypes w/ varying rates. After the cell uses the neural network to calculate p, it dies if X 〜 Binomial (p) = 1.
□ INFERNO – INFERring the molecular mechanisms of NOncoding genetic variants:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/10/30/211599.full.pdf
INFERNO provides the most comprehensive and unbiased tool to identify causal noncoding variants disrupting enhancers and the downstream effects of these disruptions including the relevant tissue context and affected target genes and pathways.
□ REQUIEM – RElative QUantitation Inferred by Evaluating Mixtures:
>>
http://www.sciencedirect.com/science/article/pii/S0003267017310723
REQUIEM is broadly applicable to diverse real-world analytical methods, including tandem mass spectrometry. the REQUIEM algorithm yields unbiased analyte fold-changes and associated statistics, allowing several types of errors to be eliminated.
□ Detecting evolutionary forces in language change:
>>
https://www.nature.com/nature/journal/vaop/ncurrent/full/nature24455.html
“デジタル化された膨大な言語資料と集団遺伝学による推論を組み合わせる”
□ The Randomness of Language Evolution:
>>
https://www.theatlantic.com/science/archive/2017/11/drove-not-drived/544595/
The understanding that gene frequencies change at random by genetic drift, even in the absence of natural selection, was a seminal advance in evolutionary biology.
□ mosdepth: quick coverage calculation for genomes and exomes
>>
https://academic.oup.com/bioinformatics/article/doi/10.1093/bioinformatics/btx699/4583630
Mosdepth uses HTSLib via the nim programming language; it expects the input BAM or CRAM file to be sorted by position. For a 5.5GB exome BAM and all 1,195,764 ensembl exons as the regions, this completes in 1 minute 38 seconds with a single CPU. The mosdepth method does require more memory – for the 249 megabase chromosome 1 in the human genome, it will require about 1GB of memory.

□ FALCON: a toolbox for the fast contextualization of logical networks:
>>
https://academic.oup.com/bioinformatics/article/doi/10.1093/bioinformatics/btx380/3897376/FALCON-A-Toolbox-for-the-Fast-Contextualisation-of
In the FALCON framework, each molecular interaction is formulated as a logical predicate associated with a weight quantifying the relative importance of that specific interaction. Hyperedges corresponding to the logical operations link multiple nodes to an output node and model the activity of complexes & competition.
□ MultiQC v1.3 released!
>>
http://multiqc.info/
7 new modules: 10X Supernova, BBMap, deepTools, Homer Tag Directory, illumina InterOp, RSEM, HiCExplorer. File searches now much faster when specifying specific modules

□ Boosting Gene Expression Clustering with System-Wide Biological Information: A Robust Autoencoder Approach:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/11/05/214122.full.pdf
an outlier filter layer before a normal autoencoder is introduced, providing robustness, while the autoencoder provides nonlinearity. LD matrix representing by a non-linear manifold, and S matrix representing the outliers which will corrupt and skew the non-linear manifold.
□ Granatum: a graphical single-cell RNA-Seq analysis pipeline for genomics scientists:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/08/08/110759.full.pdf …
The pipeline will graphically guide through the analysis of scRNA-seq data, starting from gene expression and metadata tables. Granatum has a comprehensive modules for plate merging & batch-effect removal, outlier-sample removal, filtering, normalization, clustering, DGE analysis, pathway/ontology enrichment analysis, protein-network interaction visualization, and cell pseudo-time pathway construction.
□ Arteria: An automation system for a sequencing core facility:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/11/06/214858.full.pdf
The Arteria system breaks down into three conceptual levels; orchestration, process and execution. Arteria system has been used to process more than 22000 samples and 326 projects, which corresponds to ~640 Tera-bases of sequencing data.

□ NanoSV: Mapping and phasing of structural variation in patient genomes using nanopore long-read sequencing data:
>>
https://www.nature.com/articles/s41467-017-01343-4
NanoSV uses split read mapping (obtained from LAST alignment) as a basis for SV discovery & supports discovery of all defined types of SVs. The aligned read may contain gaps between its aligned segments, i.e., parts of the read that do not align anywhere on the reference genome.

□ Integrated molecular and clinical analysis for understanding human disease relationships:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/11/06/214833.full.pdf
Gene expression meta-analysis data was compiled from the MetaSignature database. GE meta-analysis and EHR analysis, Arcs connect the same disease and clinical dendrograms and are colored based on molecular groupings.
□ K-mer clustering algorithm using a MapReduce framework: the parallelization of the Inchworm module of Trinity:
>>
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1881-8
reads from a transcribed gene yield two k-mer clusters, and hence two sets of Inchworm contigs, then the Chrysalis module should in principle find welds between them, and recover the correct graph.
□ f-scLVM: scalable and versatile factor analysis for single-cell RNA-seq:
>>
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1334-8
f-scLVM is based on a variant of sparse factor analysis, decomposing the observed gene matrix into a sum of contributions. f-scLVM was robust to extremely sparse datasets, typical of droplet-based approaches. f-scLVM more accurately identified the true simulated drivers than other methods based on PCA, linear mixed models, or factor analysis.
□ The effect of genetic variation on promoter usage and enhancer activity
>>
https://www.nature.com/articles/s41467-017-01467-7
□ Aligning sequences to general graphs in O(V + mE) time:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/11/08/216127.full.pdf
an algorithm to compute the minimum edit distance of a sequence of length m to any path in a node-labeled directed graph (V, E) in O(|V |+m|E|) time and O(|V |) space. O(mE + V m log V ) algorithm for optimally aligning a sequence to a graph with affine gap costs and arbitrary match costs.
□ LRSim: A Linked-Reads Simulator generating insights for better genome partitioning:
>>
http://www.sciencedirect.com/science/article/pii/S2001037017300855
the FALCON-Unzip algorithm and applied it to de novo assemble three phased diploid genomes. testing LRSim with the 10X Genomics LongRanger variant identification and phasing application & the 10X Genomics SuperNova genome assembler.

□ Vicus: Exploiting local structures to improve network-based analysis of biological data:
>>
http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005621
Vicus-based spectrum exhibits higher stability along the Markovian timeline compared to the global Laplacian. the Vicus matrix formulation shares algebraic properties w/ the traditional Laplacian in the task of graph-based dimensionality reduction.












 (
(