人、或いは集団の意思決定プロセスを分子軌道から決定論的に解析出来るとしたら、それはまるで相転移する漣のようなモアレを描くはずである。
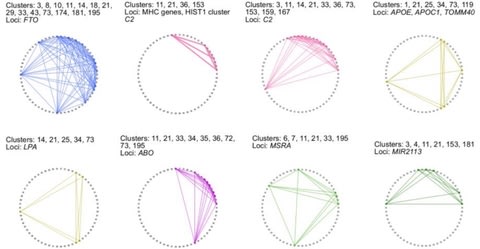
□ Phenome-wide search for pleiotropic loci highlights key genes and molecular pathways for human complex traits
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/16/672758.full.pdf
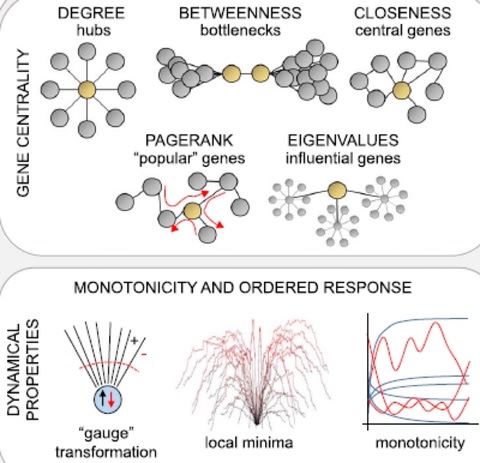
Pleiotropy of trait-associated variants in the human genome has also attracted lots of attention in the field; and Mendelian randomization based approaches have been proposed to detect pleiotropy in GWAS data.
a statistical framework to explore the landscape of phenome-wide associations in GWAS summary statistics derived from UK Biobank dataset, and identified multiple shared blocks of genetic architecture of diverse human complex traits.

□ These Sumptuous Images Give Deep Space Data An Old-World look
>> https://www.wired.com/story/these-sumptuous-images-give-deep-space-data-an-old-world-look/
Eleanor Lutz is a biologist with a knack for producing visually rich data visualizations. She's done everything from animated viruses to infographics on plant species that have evolved to withstand forest fires.
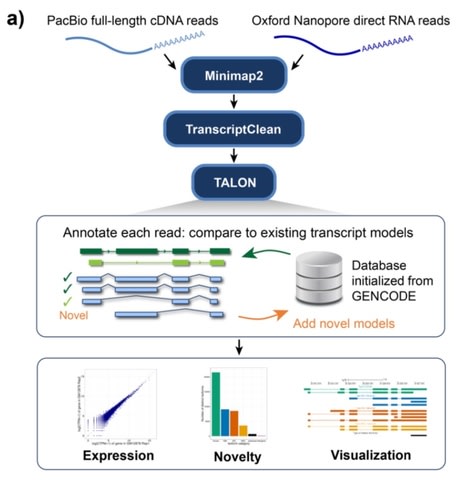
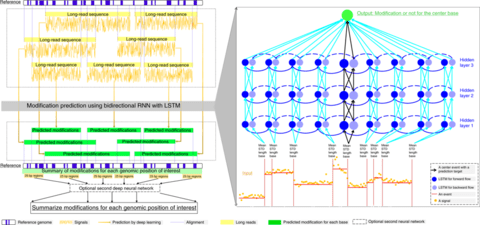
□ TALON: A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/18/672931.full.pdf
TALON is the ENCODE4 pipeline for analyzing PacBio cDNA and ONT direct-RNA transcriptomes.
TALON pipeline for technology-agnostic, long-read transcriptome discovery and quantification tracks both known and novel transcript models as well as expression levels across datasets for both simple studies and larger projects such that seek to decode transcriptional regulation.

□ scEntropy: Single-cell entropy to quantify the cellular transcriptome from single-cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/21/678557.full.pdf
scEntropy can be considered as a one-dimensional stochastic neighbour embedding of the original data.
the use of single-cell entropy (scEntropy) to measure the order of the cellular transcriptome profile from single-cell RNA-seq data, which leads to a method of unsupervised cell type classification through scEntropy followed by the Gaussian mixture model (scEGMM).
the idea of finding the entropy of a system with reference to a baseline can easily be generalized to other applications, which extends the classical concepts of entropy in describing complex systems.
□ DEUS: an R package for accurate small RNA profiling based on differential expression of unique sequences
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz495/5522007
DEUS is a novel profiling strategy that circumvents the need for read mapping to a reference genome by utilizing the actual read sequences to determine expression intensities.
After differential expression analysis of individual sequence counts, significant sequences are annotated against user defined feature databases and clustered by sequence similarity.
DEUS strategy enables a more comprehensive and concise representation of small RNA populations without any data loss or data distortion.
□ ivis: Structure-preserving visualisation of high dimensional single-cell datasets
>> https://www.nature.com/articles/s41598-019-45301-0
ivis is a novel framework for dimensionality reduction of single-cell expression data.
ivis utilizes a siamese neural network architecture that is trained using a novel triplet loss function. Each triplet is sampled from one of the k nearest neighbours as approximated by the Annoy library, neighbouring points being pulled together & non-neighours being pushed away.
ivis learns a parametric mapping from the high-dimensional space to low-dimensional embedding, facilitating seamless addition of new data points to the mapping function.

□ Aneuvis: web-based exploration of numerical chromosomal variation in single cells
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2842-1
Aneuvis is allows users to determine whether numerical chromosomal variation exists between experimental treatment groups.
Aneuvis operates downstream of existing experimental and computational approaches that generate a matrix containing the estimated chromosomal copy number per cell.
□ LAVENDER: latent axes discovery from multiple cytometry samples with non-parametric divergence estimation and multidimensional scaling reconstruction
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/18/673434.full.pdf
a computational method termed LAVENDER (latent axes discovery from multiple cytometry samples with nonparametric divergence estimation and multidimensional scaling reconstruction).
Jensen-Shannon distances between samples using the k-nearest neighbor density estimation and reconstructs samples in a new coordinate space, called the LAVENDER space.
□ MetaCurator: A hidden Markov model-based toolkit for extracting and curating sequences from taxonomically-informative genetic markers
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/18/672782.full.pdf
Aside from modules used to organize and format taxonomic lineage data, MetaCurator contains two signature tools.
IterRazor utilizes profile hidden Markov models and an iterative search framework to exhaustively identify and extract the precise amplicon marker of interest from available reference sequence data.

□ NAGA: A Fast and Flexible Framework for Network-Assisted Genomic Association
>> https://www.cell.com/iscience/fulltext/S2589-0042(19)30162-2
NAGA (Network Assisted Genomic Association)—taps the NDEx biological network resource to gain access to thousands of protein networks and select those most relevant and performative for a specific association study.
NAGA is based on the method of network propagation, which has emerged as a robust and widely used network analysis technique in many bioinformatics applications.
PEGASUS finds an analytical model for the expected chi-square statistics because of correlation from linkage disequilibrium, which worked well with the network propagation algorithm.

□ Modular and efficient pre-processing of single-cell RNA-seq
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/17/673285.full.pdf
a Chromium pre-processing workflow based on reasoned choices for the key pre-processing steps.
this workflow is based on the kallisto and bustools programs, and is near-optimal in speed and memory.
This scRNA-seq workflow is up to 51 times faster than Cell Ranger and up to 4.75 times faster than Alevin. It is also up to 3.5 times faster than STARsolo: a recent version of the STAR aligner.
identical UMIs associated with distinct reads from the same gene are almost certainly reads from the same molecule, makes it possible, in principle, to design efficient assignment algorithms for multi-mapping reads.
Distinct technology encodes barcode and UMI information differently in reads, but the kallisto bus command can accept custom formatting rules.
□ SMNN: Batch Effect Correction for Single-cell RNA-seq data via Supervised Mutual Nearest Neighbor Detection
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/17/672261.full.pdf
SMNN either takes cluster/cell-type label information as input or infers cell types using scRNA-seq clustering in the absence of such information.
It then detects mutual nearest neighbors within matched cell types and corrects batch effect accordingly.

□ NanoVar: Accurate Characterization of Patients' Genomic Structural Variants Using Low-Depth Nanopore Sequencing
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/17/662940.full.pdf
NanoVar, an accurate, rapid and low-depth (4X) 3GS SV caller utilizing long-reads generated by Oxford Nanopore Technologies.
NanoVar demonstrated the highest SV detection accuracy (F1 score = 0.91) amongst other long-read SV callers using 12 gigabases (4X) of sequencing data.
NanoVar employs split-reads and hard-clipped reads for SV detection and utilizes a neural network classifier for true SV enrichment.

□ A multimodal framework for detecting direct and indirect gene-gene interactions from large expression compendium
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/23/680116.full.pdf
a Multimodal framework (MMF) to depict the gene expression profiles. MMF introduces two new statistics: Multimodal Mutual Information and Multimodal Direct Information.
In the principal component analysis for very large collections of expression data, the use of Multimodal Mutual Information (MMI) enables more biologically meaningful spaces to be extracted than the use of Pearson correlation.
Multimodal Direct Information, which is enhanced from MMI based on maximum entropy principle.
□ High-throughput multiplexed tandem repeat genotyping using targeted long-read sequencing:
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/17/673251.full.pdf
Genotyping estimates from targeted long-read sequencing were determined using two different methods (VNTRTyper and Tandem-genotypes) and results were comparable.
Furthermore, genotyping estimates from targeted long-read sequencing were highly correlated with genotyping estimates from whole genome long-read sequencing.

□ Duplication-divergence model (DD-model): Revisiting Parameter Estimation in Biological Networks: Influence of Symmetries
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/18/674739.full.pdf
a parameter estimation scheme for biological data with a new perspective of symmetries and recurrence relations, and point out many fallacies in the previous estimation procedures.
Parameter estimation provides us with better knowledge about the specific characteristics of the model that retains temporal information in its structure.
Since the inference techniques are closely coupled with the arrival process, assuming that networks evolve according to the duplication-divergence stochastic graph model.
□ Bayesian inference of power law distributions
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/18/664243.full.pdf
BayesPowerlaw fits single or mixtures of power law distributions and estimate their exponent using Bayesian Inference, specifically Markov-Chain Monte Carlo Metropolis Hastings algorithm.
a probabilistic solution to these issues by developing a Bayesian inference approach, with Markov chain Monte Carlo sampling, to accurately estimate power law exponents, the number of mixtures, and their weights, for both discrete and continuous data.
□ MetaSeek: Sequencing Data Discovery
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz499/5521620
MetaSeek scrapes metadata from the sequencing data repositories, cleaning and filling in missing or erroneous metadata, and stores the cleaned and structured metadata in the MetaSeek database.
MetaSeek automatically scrapes metadata from all publicly available datasets in the Sequence Read Archive, cleans and parses messy, user-provided metadata into a structured, standard-compliant database, and predicts missing fields where possible.
□ Deep Learning on Chaos Game Representation for Proteins
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz493/5521624
using frequency matrix chaos game representation (FCGR) for encoding of protein sequences into images.
While the original chaos game representation (CGR) has been used mainly for genome sequence encoding and classification, modifying it to work also for protein sequences, resulting in n-flakes representation, an image with several icosagons.
□ A Multidimensional Array Representation of State-Transition Model Dynamics
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/21/670612.full.pdf
modifying the transitional cSTMs cohort trace computation to compute and store cSTMs dynamics that capture both state occupancy and transition dynamics.
This approach produces a multidimensional matrix from which both the state occupancy and the transition dynamics can be recovered.

□ SPsimSeq: semi-parametric simulation of bulk and single cell RNA sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/21/677740.full.pdf
SPsimSeq simulates data from a good estimate of the actual distribution of a given real RNA-seq dataset.
In contrast to existing approaches that assume a particular data distribution, SPsimSeq constructs an empirical distribution of gene expression data from a given source RNA-seq experiment to faithfully capture the data characteristics of real data.
SPsimSeq can be used to simulate a wide range of scenarios, such as single or multiple biological groups, systematic variations (e.g. confounding batch effects), and different sample sizes.
SPsimSeq can also be used to simulate different gene expression units resulting from different library preparation protocols, such as read counts or UMI counts.
□ LR_EC_analyser: Comparative assessment of long-read error correction software applied to Nanopore RNA-sequencing data
>> https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbz058/5512144
an automatic and extensive benchmark tool that not only reports classical error correction metrics but also the effect of correction on gene families, isoform diversity, bias toward the major isoform and splice site detection.
long read error correction tools that were originally developed for DNA are also suitable for the correction of Nanopore RNA-sequencing data, especially in terms of increasing base pair accuracy.
LR_EC_analyser can be applied to evaluate the extent to which existing long-read DNA error correction methods are capable of correcting long reads.
□ Denseness conditions, morphisms and equivalences of toposes
>> https://arxiv.org/pdf/1906.08737v1.pdf
a general theorem providing necessary and sufficient explicit conditions for a morphism of sites to induce an equivalence of toposes.
This results from a detailed analysis of arrows in Grothendieck toposes and denseness conditions, which yields results of independent interest.
And also derive site characterizations of the property of a geometric morphism to be an inclusion (resp. a surjection, hyper-connected, localic), as well as site-level descriptions of the surjection- inclusion and hyperconnected-localic factorizations.
□ PaKman: Scalable Assembly of Large Genomes on Distributed Memory Machines
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/17/523068.full.pdf
PaKman presents a solution for the two most time-consuming phases in the full genome assembly pipeline, namely, k-mer counting and contig generation.
PaKman is able to generate a high-quality set of assembled contigs for complex genomes such as the human and wheat genomes in a matter of minutes on 8K cores.
A key aspect of this algorithm is its graph data structure, which comprises fat nodes (or what we call “macro-nodes”) that reduce the communication burden during contig generation.
□ Global-and-local-structure-based neural network for fault detection
>> https://www.sciencedirect.com/science/article/pii/S0893608019301625
GLSNN is a nonlinear data-driven process monitoring technique through preserving both global and local structures of normal process data.
GLSNN is characterized by adaptively training a neural network which takes both the global variance information and the local geometrical structure into consideration.

□ Manatee: detection and quantification of small non-coding RNAs from next-generation sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/22/662007.full.pdf
sMAll rNa dATa analysis pipElinE (MANATEE) achieves highly accurate results, even for elements residing in heavily repeated loci, by making balanced use of existing sRNA annotation and observed read density information during multi-mapper placement.
Manatee adopts a novel approach for abundance estimation of genomic reads that combines sRNA annotation with reliable alignment density information and extensive reads salvation.
□ fpmax: Maximal Itemsets via the FP-Max Algorithm
>> https://github.com/rasbt/mlxtend/blob/master/docs/sources/user_guide/frequent_patterns/fpmax.ipynb
In contrast to Apriori, FP-Growth is a frequent pattern generation algorithm that inserts items into a pattern search tree, which allows it to have a linear increase in runtime with respect to the number of unique items or entries.
FP-Max is a variant of FP-Growth, which focuses on obtaining maximal itemsets. An itemset X is said to maximal if X is frequent and there exists no frequent super-pattern containing X.
a frequent pattern X cannot be sub-pattern of larger frequent pattern to qualify for the definition maximal itemset.
□ New York Genome Center awarded $1.5M CZI grant for single-cell analysis toolkit
>> https://eurekalert.org/pub_releases/2019-06/nygc-nyg062119.php
Multi-Modal Cell Profiling and Data Integration to Atlas the Immune System is the collaborative project will take advantage of the strengths of each of the core technologies: multimodal RNA data from CITE-seq, developed in the NYGC Technology Innovation Lab.
□ An explicit formula for a dispersal kernel in a patchy landscape
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/23/680256.full.pdf
Integrodifference equations (IDEs) are often used for discrete-time continuous-space models in mathematical biology.
derive a generalization of the classic Laplace kernel, which includes different dispersal rates in each patch as well as different degrees of bias at the patch boundaries.
an explicit formula for the kernel as piecewise exponential function with coefficients and rates determined by the inverse of a matrix of model parameters.
□ Performance of neural network basecalling tools for Oxford Nanopore sequencing:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1727-y
Albacore, Guppy and Scrappie all use an architecture that ONT calls RGRGR – named after its alternating reverse-GRU and GRU layers.
To test whether more complex networks perform better, modify ONT’s RGRGR network by widening the convolutional layer and doubling the hidden layer size.
Chiron is a third-party basecaller still under development that uses a deeper neural network than ONT’s basecallers. Chiron v0.3 had the highest consensus accuracy (Q25.9) of all tested basecallers using their default models.

□ Biomine Explorer: Interactive exploration of heterogeneous biological networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz509/5522368
Biomine Explorer enables interactive exploration of large heterogeneous biological networks constructed from selected publicly available biological knowledge sources.
It is built on top of Biomine, a system which integrates cross-references from several biological databases into a large heterogeneous probabilistic network.
□ MaNGA: a novel multi-objective multi-niche genetic algorithm for QSAR modelling
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz521/5522367
a new multi-niche/multi-objective genetic algorithm (MaNGA) that simultaneously enables stable feature selection as well as obtaining robust and validated regression models with maximized applicability domain.
This algorithm is a valid alternative to classical QSAR modelling strategy, for continuous response values, since it automatically finds the model w/ the best compromise b/w statistical robustness, predictive performance, widest AD, & the smallest number of molecular descriptors.
□ PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz469/5522366
a major update of PhenoScanner, incl over 150 million genetic variants and more than 65 billion associations with diseases and traits, gene expression, metabolite and protein levels, and epigenetic markers.
The query options have been extended to include searches by genes, genomic regions and phenotypes, as well as for genetic variants. All variants are positionally annotated using the Variant Effect Predictor and the phenotypes are mapped to Experimental Factor Ontology terms.
Linkage disequilibrium statistics from the 1000 Genomes project can be used to search for phenotype associations with proxy variants.
□ NPCMF: Nearest Profile-based Collaborative Matrix Factorization method for predicting miRNA-disease associations
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2956-5
When novel MDAs are predicted, the nearest neighbour information for miRNAs and diseases is fully considered.
incorporating the Gaussian interaction profile kernels of miRNAs and diseases also contributed to the improvement of prediction performance.
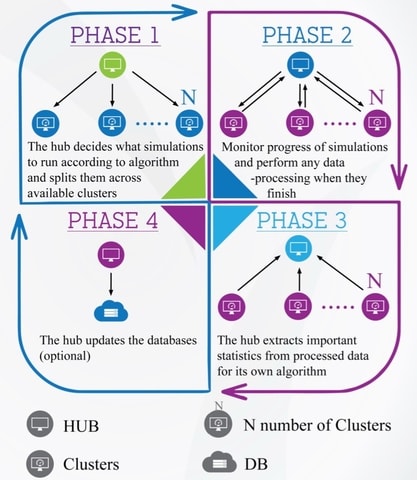
□ PyGDS: The genome design suite: enabling massive in-silico experiments to design genomes
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/24/681270.full.pdf
PyGDS provides a framework with which to implement phenotype optimisation algorithms on computational models across computer clusters.
The framework is abstract allowing it to be adapted to utilise different computer clusters, optimisation algorithms, or design goals.
It implements an abstract multi-generation algorithm structure allowing algorithms to avoid maximum simulation times on clusters and enabling iterative learning in the algorithm.

□ Deriving Disease Modules from the Compressed Transcriptional Space Embedded in a Deep Auto-encoder
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/24/680983.full.pdf
The hypothesis is that such modules could be discovered in the deep representations within the auto-encoder when trained to capture the variance in the input-output map of the transcriptional profiles.
Using a three-layer deep auto-encoder we find a statistically significant enrichment of GWAS relevant genes in the third layer, and to a successively lesser degree in the second and first layers respectively.
using deep AE with a subsequent knowledge-based interpretation scheme, enables systems medicine to become sufficiently powerful to allow unbiased identification of complex novel gene-cell type interactions of relevance for realizing systems medicine.
□ Ultraplexing: Increasing the efficiency of long-read sequencing for hybrid assembly with k-mer-based multiplexing
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/24/680827.full.pdf
Ultraplexing uses inter-sample genetic variability, as measured by Illumina sequencing, to assign long reads to individual isolates.
Ultraplexing-based assemblies are highly accurate in terms of genome structure and consensus accuracy and exhibit quality characteristics comparable to assemblies based on molecular barcoding.

□ TGAC Browser: an open-source genome browser for non-model organisms
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/24/677658.full.pdf
the TGAC Browser, a genome browser that relies on non-proprietary software but only readily available Ensembl Core database and NGS data formats.
□ Generating high-quality reference human genomes using PromethION nanopore sequencing
>> https://www.slideshare.net/MitenJain/generating-highquality-reference-human-genomes-using-promethion-nanopore-sequencing
□ PICKER-HG: a web server using random forests for classifying human genes into categories
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/24/681460.full.pdf
a PerformIng Classification and Knowledge Extraction via Rules using random forests on Human Genes (PICKER-HG), dynamically constructs a classification dataset, given a list of human genes with annotations entered by the user, and outputs classification rules extracted of a Random Forest model.