“We shall not cease from exploration
And the end of all our exploring
Will be to arrive where we started
And know the place for the first time.”
- T.S. Eliot, Four Quartets
“If they were complicated enough, both sides could sustain observers who would perceive time going in opposite directions. Any intelligent beings there would define their arrow of time as moving away from this central state. They would think we now live in their deepest past." -Julian Barbour
GARFIELD is a novel approach that leverages genome-wide association studies’ findings with regulatory or functional annotations to classify features relevant to a phenotype of interest. they assess enrichment of genome-wide association studies for 19 traits within Encyclopedia of DNA Elements- and Roadmap-derived regulatory regions. GARFIELD uncovered statistically significant enrichments for the majority of traits being considered, and highlighted clear differences in enrichment patterns between traits.
□ Apollo: A Sequencing-Technology-Independent, Scalable, and Accurate Assembly Polishing Algorithm:
Apollo, a universal assembly polishing algorithm that is scalable to polish an assembly of any size with reads from all sequencing technologies. Apollo models an assembly as a profile hidden Markov model (pHMM), uses read- to-assembly alignment to train the pHMM with the Forward-Backward algorithm, and decodes the trained model with the Viterbi algorithm to produce a polished assembly.
□ Skyhawk: An Artificial Neural Network-based discriminator for reviewing clinically significant genomic variants:
Skyhawk, an artificial neural network-based discriminator that mimics the process of expert review on clinically significant genomics variants. Among the false positive singletons identified by GATK HaplotypeCaller, UnifiedGenotyper and 16GT in the HG005 GIAB sample, 79.7% were rejected by Skyhawk.
Skyhawk mimics how a human visually identifies genomic features comprising a variant and decides whether the evidence supports or contradicts the sequencing read alignments. Skyhawk repurposed the network architecture they developed in a previous study named Clairvoyante.
□ SORA: Scalable Overlap-graph Reduction Algorithms for Genome Assembly using Apache Spark in the Cloud:
SORA adapts string graph reduction algorithms for the genome assembly using a distributed computing platform. To efficiently compute coverage for enormous paths, useing Apache Spark which is a cluster-based engine designed on top of Hadoop to handle large datasets in the cloud. The results show that SORA can process a nearly one billion edge graph in a distributed cloud cluster as well as smaller graphs on a local cluster with a short turnaround time. Their algorithms scale almost linearly with increasing numbers of virtual instances in the cloud.
□ CONSENT: Scalable self-correction of long reads with multiple sequence alignment:
CONSENT (sCalable self-cOrrectioN of long reads with multiple SEquence alignmeNT) is a self-correction method for long reads. It works by computing overlaps b/n the long reads, in order to define an alignment pile (a set of overlapping reads used for correction) for each read. CONSENT compares well to the latest state-of-the-art self-correction methods, and even outperforms them on real Oxford Nanopore datasets. CONSENT is the only method able to scale to a human dataset containing Oxford Nanopore ultra-long reads, reaching lengths up to 340 kbp.
□ Fast and accurate long-read assembly with wtdbg2:
a novel long-read assembler wtdbg2 that, for human data, is tens of times faster than published tools while achieving comparable contiguity and accuracy. Wtdbg2 broadly follows the overlap-layout-consensus paradigm. It advances the existing assemblers with a fast all-vs-all read alignment implementation and a novel layout algorithm based on fuzzy-Bruijn graph (FBG).
Wtdbg2 bins read sequences to speed up the next step in alignment: dynamic programming (DP). With 256bp binning, the DP matrix is 65536 (=256 ́256) times smaller than a per-base DP matrix. For all human data, wtdbg2 finishes the assembly in a few days on a single computer. This performance broadly matches the throughput of a PromethION machine.
□ RUV-z: A causal inference framework for estimating genetic variance and pleiotropy from GWAS summary data:
RUV-z (Removing Unwanted Variation in GWAS z-score matrix), with which we characterize undesired sources of information lurking in summary statistics, and selectively remove them to improve accuracy and statistical power of local variance/covariance calculation. zQTL (z-score based quantitative trait locus analysis), a suite of machine learning methods for summary-based regression and matrix factorization, then demonstrate how they can successively apply the factorization and regression steps to design a new confounder-correction method.
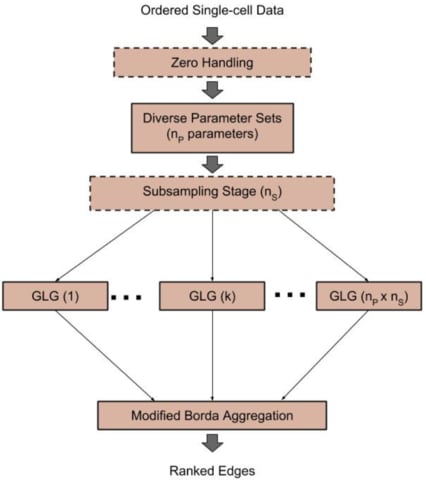
□ SCINGE: Network Inference with Granger Causality Ensembles on Single-Cell Transcriptomic Data:
Single-Cell Inference of Networks using Granger Ensembles (SCINGE) algorithm, an ensemble-based GRN reconstruction tech- nique that uses modified Granger Causality on single-cell data annotated with pseudotimes. Within SCINGE, GLG uses a kernel function to smooth the past expression values of candidate regulators, mitigating the irregularly-spaced pseudotimes and zero values that are prevalent in single- cell expression data.
SCINGE compares favorably with existing GRN inference methods designed for temporal or pseudotemporal GE data. it reveals important caveats about GRN evaluation and the value of pseudotime for GRN inference that are broadly applicable for pseudotime-based GRN reconstruction.
The goal of causal network reconstruction or causal discovery is to distinguish direct from indirect dependencies and common drivers among multiple time series. A variety of different assumptions have been shown to be sufficient to estimate the true causal graph. focussing on three main assumptions under which the time series graph represents causal relations: Causal Sufficiency, the Causal Markov Condition, and Faithfulness.
□ USDL: A Unified Approach for Sparse Dynamical System Inference from Temporal Measurements:
Unified Sparse Dynamics Learning (USDL), constitutes of two steps. First, an atemporal system of equations is derived through the application of the weak formulation. Then, assuming a sparse representation for the dynamical system, the inference problem can be expressed as a sparse signal recovery problem, allowing the application of an extensive body of algorithms and theoretical results.
Results on simulated data demonstrate the efficacy and superiority of the USDL algorithm under multiple interventions and/or stochasticity. Additionally, USDL’s accuracy significantly correlates with theoretical metrics such as the exact recovery coefficient. On real single-cell data, the proposed approach is able to induce high-confidence subgraphs of the signaling pathway.
□ LuxUS: Detecting differential DNA methylation using generalized linear mixed model with spatial correlation structure:
LuxGLM Using Spatial correlation (LuxUS) is a tool for differential methylation analysis. The tool is based on generalized linear mixed model with spatial correlation structure. The model parameters are fitted using probabilistic programming language Stan. Savage-Dickey Bayes factor estimates are used for statistical testing of a covariate of interest. LuxUS supports both continuous and binary variables. The model takes into account the experimental parameters, such as bisulfite conversion efficiency.
catch22 captures a diverse and interpretable signature of time series in terms of their properties, including linear and non-linear autocorrelation, successive differences, value distributions and outliers, and fluctuation scaling properties. For 85 of the 93 datasets, unbalanced classification accuracies were provided for different shape-based classifiers such as dynamic time warping (DTW) nearest neighbor, as well as for hybrid approaches such as COTE.
□ Bayesian Multiple Emitter Fitting using Reversible Jump Markov Chain Monte Carlo:
a Bayesian inference approach to multiple- emitter fitting that uses Reversible Jump Markov Chain Monte Carlo to identify and localize the emitters in dense regions of data. The output is both a posterior probability distribution of emitter locations that includes uncertainty in the number of emitters and the background structure, and a set of coordinates and uncertainties from the most probable model.
□ scVI/scANVI: Harmonization and Annotation of Single-cell Transcriptomics data with Deep Generative Models:
scANVI, a semi-supervised variant of scVI designed to leverage any available cell state annotations — for instance when only one data set in a cohort is annotated, or when only a few cells in a single data set can be labeled using marker genes. scVI and scANVI methods provide a complete probabilistic representation of the data, which non-linearly controls not only for sample-to-sample bias but also for other technical factors of variation such as over-dispersion, library size discrepancies and zero-inflation.
□ Re-curation and Rational Enrichment of Knowledge Graphs in Biological Expression Language:
A workflow for re-curating and rationally enriching knowledge graphs encoded in Biological Expression Language using pre-extracted content from INDRA. Furthermore, INDRA is flexible enough to generate curation sheets for curators familiar with formats other than BEL, such as BioPAX or SBML.
□ Computational analysis of molecular networks using spectral graph theory, complexity measures and information theory:
Spectral graph theory, reciprocal link and complexity measures were utilized to quantify network motifs. It was found that graph energy, reciprocal link and cyclomatic complexity can optimally specify network motifs with some degree of degeneracy. Biological networks are built up from a finite number of motif patterns; hence, a graph energy cutoff exists and the Shannon entropy of the motif frequency distribution is not maximal. Network similarity was quantified by gauging their motif frequency distribution functions using Jensen-Shannon entropy. This method allows us to determine the distance between two networks regardless of their node identities and network sizes.
□ SuperCRUNCH: A toolkit for creating and manipulating supermatrices and other large phylogenetic datasets:
SuperCRUNCH can be used to generate interspecific supermatrix datasets (one sequence per taxon per locus) or population-level datasets (multiple sequences per taxon per locus). It can also be used to assemble phylogenomic datasets with thousands of loci.
□ Simulating the DNA String Graph in Succinct Space:
rBOSS is a de Bruijn graph in practice, but it simulates any length up to k and can compute overlaps of size at least m between the labels of the nodes, with k and m being parameters. As most BWT-based structures, rBOSS is unidirectional, but it exploits the property of the DNA reverse complements to simulate bi-directionality with some time-space trade-offs.
□ Garnett: Supervised classification enables rapid annotation of cell atlases
Garnett, an algorithm and accompanying software for rapidly annotating cell types in scRNA-seq and scATAC-seq datasets, based on an interpretable, hierarchical markup language of cell type-specific genes. Garnett will expand classifications to similar cells to generate a separate set of cluster-extended type assignments. Garnett successfully classifies cell types in tissue and whole organism datasets, as well as across species.
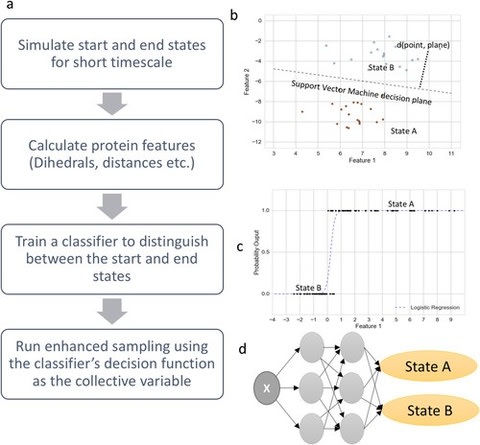
□ Automated design of collective variables using supervised machine learning:
SMLCV shows how the distance to the support vector machines’ decision hyperplane, the output probability estimates from logistic regression, the outputs from deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions.
a theoretical framework that describes conditions under which reservoir computing can create an empirical model capable of skillful short-term forecasts and accurate long-term ergodic behavior. a theory of how prediction with discrete-time reservoir computing or related machine-learning methods can “learn” a chaotic dynamical system well enough to reconstruct the long-term dynamics of its attractor.
□ Isospectral deformations, the spectrum of Jacobi matrices, infinite continued fraction and difference operators. Application to dynamics on infinite dimensional systems:
The use of tau functions related to infinite dimensional Grassmannians, Fay identities, vertex operators and the Hirota’s bilinear formalism led to obtaining important results concerning these algebras of infinite order differential operators. In addition many problems related to algebraic geometry, combinatorics, probabilities and quantum gauge theory,..., have been solved explicitly by methods inspired by techniques from the study of dynamical integrable systems.
Heterogeneous quantifiers (infinite alternations of universal and existential quantification) present a new kind of quantification in infinitary logic related to game semantics. a proof system for classical infinitary logic that includes heterogeneous quantification (i.e., infinite alternate sequences of quantifiers) within the language Lκ+,κ. In κ-Grothendieck toposes in particular, and, when κ<κ = κ, also in Kripke models.
□ Grid-LMM: Fast and flexible linear mixed models for genome-wide genetics:
Grid-LMM, an extendable algorithm for repeatedly fitting complex linear models that account for multiple sources of heterogeneity, such as additive and non-additive genetic variance, spatial heterogeneity, and genotype-environment interactions. Grid-LMM includes functions for both frequentist and Bayesian GWAS, (Restricted) Maximum Likelihood evaluation, Bayesian Posterior inference of variance components, and Lasso/Elastic Net fitting of high-dimensional models with random effects.
Supposedly “uniformative” versions of both the inverse-Gamma and half-Cauchy-type priors are actually highly informative for variance component proportions. A uniform prior over the grid was assumed, and the intercept was assigned a Gaussian prior with infinite variance.
□ hilldiv: an R package for the integral analysis of diversity based on Hill numbers:
Hill numbers provide a powerful framework for measuring, estimating, comparing and partitioning the diversity of biological systems as characterised using high throughput DNA sequencing approaches. The statistical framework developed around Hill numbers encompasses many of the most broadly employed diversity (e.g. richness, Shannon index, Simpson index), phylogenetic diversity (quadratic entropy) and dissimilarity (e.g. Sørensen index, Unifrac distances) metrics.
□ cSG-MCMC: Cyclical Stochastic Gradient MCMC for Bayesian Deep Learning:
Several attempts have been made to improve the sampling efficiency of SG-MCMC. Stochastic Gradient Hamiltonian Monte Carlo (SGHMC) introduces the momentum variable to the Langevin dynamics.
the posteriors over neural network weights are high dimensional and multimodal. Each mode typically characterizes a meaningfully different representation of the data. Cyclical Stochastic Gradient MCMC (SG-MCMC) automatically explore such distributions. cyclical SG-MCMC methods provide more accurate uncertainty estimation, by capturing more diversity in the hypothesis space corresponding to settings of model parameters.
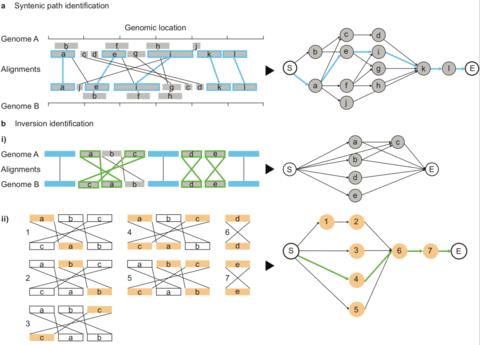
□ SyRI: identification of syntenic and rearranged regions from whole-genome assemblies:
Any pair of nodes is then connected by an edge if the two underlying alignments are colinear. Alignments are defined as colinear if the underlying regions are non-rearranged to each other and if no other co-linear alignment is between them. SyRI identifies the maximal syntenic path (i.e. the optimal set of non-conflicting, co-linear regions) by selecting the highest scoring path between node S (Start) and E (End) using dynamic programming.
□ BLight: Indexing De Bruijn graphs with minimizers:
BLight is a scalable and exact index structure able to associate unique identifiers to indexed k-mers and to reject alien k-mers. The proposed structure combines an extremely compact representation along with a high throughput. BLight, an ubiquitous, efficient and exact associative structure for indexing k-mers, relying on De Bruijn graphs. Based on efficient hashing techniques and light memory structure.
a new method for high-throughput sequencing of polyadenylated RNAs in their entirety, including the transcription start site, the splicing pattern, the 3’ end and the poly(A) tail for each sequenced molecule. By providing full-length mRNA sequence including the poly(A) tail, FLAM-seq allows to reconstruct dependencies between different levels of gene regulation - in particular promoter choice, alternative splicing, 3’ UTR choice, and polyA tail length.
□ MIA: Andrew Blumberg, Using random matrix theory to model single-cell RNA; topological data analysis
a method for low-rank approximation of a data matrix arising from single-cell RNA sequencing data. The basic observation is that such data is consistent with a sparse version of the "spike model" studied in random matrix theory.
□ Network inference performance complexity: a consequence of topological, experimental, and algorithmic determinants:
conducted a multifactorial analysis that demonstrates how stimulus target, regulatory kinetics, induction and resolution dynamics, and noise differentially impact widely used algorithms in significant and previously unrecognized ways. The results show how even if high-quality data are paired with high-performing algorithms, inferred models are sometimes susceptible to giving misleading conclusions.
□ Scalable multiple whole-genome alignment and locally collinear block construction with SibeliaZ:
SibeliaZ-LCB for identifying collinear blocks in closely related genomes based on the analysis of the de Bruijn graph. SibeliaZ shows drastic run-time improvements over other methods on both simulated and real data, with only a limited decrease in accuracy. SibeliaZ works by first constructing the compacted de Bruijn graph using our previoulsy published TwoPaCo tool, then finding locally collinear blocks using using SibeliaZ-LCB, and finally, running a multiple-sequence aligner on each of the found blocks.
□ SCENT: Estimating Differentiation Potency of Single Cells Using Single-Cell Entropy:
The estimation of differentiation potency is based on an explicit biophysical model that integrates the RNA-Seq profile of a single cell with an interaction network to approximate potency as the entropy of a diffusion process on the network.
□ Kevlar: a mapping-free framework for accurate discovery of de novo variants:
Kevlar identifies high-abundance k-mers unique to the individual of interest and retrieves the reads containing these k-mers. These reads are easily partitioned into disjoint sets by shared k-mer content for subsequent locus-by-locus processing and variant calling. Kevlar employs a novel probabilistic model to score variant predictions and distinguish miscalled inherited variants and true de novo mutations.
kevla predicts de novo genetic variants without mapping reads to a reference genome. kevlar's k-mer abundance based method calls single nucleotide variants, multinucleotide variants, insertion/deletion variants, and structural variants simultaneously with a single simple model.
□ Compositional Data Network Analysis via Lasso Penalized D-Trace Loss:
A sparse matrix estimator for the direct interaction network is defined as the minimizer of lasso penalized CD-trace loss under positive-definite constraint. Simulation results show that CD-trace compares favorably to gCoda and that it is better than sparse inverse covariance estimation for ecological association inference (SPIEC-EASI) (hereinafter S-E) in network recovery with compositional data.
□ Virtual ChIP-seq: Predicting transcription factor binding by learning from the transcriptome:
Virtual ChIP-seq, which predicts binding of individual transcription factors in new cell types using an artificial neural network that integrates ChIP-seq results from other cell types and chromatin accessibility data in the new cell type. Virtual ChIP-seq also uses learned associations between gene expression and transcription factor binding at specific genomic regions.
The main purpose is to introduce the base system RCA0 which stands for Recursive Comprehesion Axiom system, and which is still missing the concept of general computable sets to actually prove theorems like for example the Heine-Borel theorem in the stronger ACA0 system.
if a finitely branching tree has infinitely many vertices, then it has an infinite path. Its proof resembles the proof of the Bolzano-Weierstrass or the Heine-Borel theorem that rely on the construction of an infinite sequence of nested intervals.
All these proofs incorporate an enumerable concept that should result in the definition of limit.
ProteinNet integrates sequence, structure, and evolutionary information in programmatically accessible file formats tailored for machine learning frameworks. Multiple sequence alignments of all structurally characterized proteins were created using substantial high-performance computing resources. Standardized data splits were also generated to emulate the difficulty of past CASP (Critical Assessment of protein Structure Prediction) experiments by resetting protein sequence and structure space to the historical states that preceded six prior CASPs.
□ Pyramid Model: A general framework for moment-based analysis of genetic data:
a novel Hierarchical Beta approximation is developed, a Pyramidal Hierarchical Beta model, for the generalized time-reversible and single-step mutation processes.
the validity of the Dirichlet distribution has never been systematically investigated in a general framework. Attempt to address this problem by providing a general overview of how allele fraction data under the most common multi-allelic mutational structures should be modeled. The Dirichlet and alternative models are investigated by simulating allele fractions from a diffusion approximation of the multi-allelic Wright–Fisher process with mutation, and applying a moment-based analysis method.
The Dirichlet model is only an exceptionally good approximation for the pure drift, Jukes–Cantor and parent-independent mutation processes with small mutation rates. Alternative models are required and proposed for the other mutation processes, such as a Beta–Dirichlet model for the infinite alleles mutation process, and a Hierarchical Beta model.
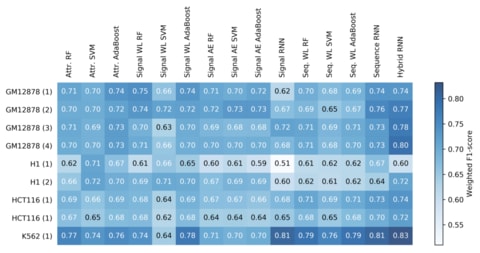
□ ATAC-seq signal processing and recurrent neural networks can identify RNA polymerase activity:
a frequency space analysis of the “signal” generated from ATAC-seq experiments’ short reads, at a single-nucleotide resolution, using a discrete wavelet transform. analyzing the performance of different data encoding schemes and machine learning architectures, and show how a hybrid signal/sequence representation classified using recurrent neural networks (RNNs) yields the best performance across different cell types.
□ From expression footprints to causal pathways: contextualizing large signaling networks with CARNIVAL:
CARNIVAL (CAusal Reasoning pipeline for Network identification using Integer VALue programming) integrates different sources of prior knowledge, including signed and directed protein-protein interactions, transcription factor targets, and pathway signatures. CARNIVAL allows the capture of a broad set of upstream cellular processes & regulators, which in turn delivered results w/ higher accuracy when benchmarked against related tools. Implementation as an integer linear programming (ILP) problem also guarantees efficient computation.
□ VEF: a Variant Filtering tool based on Ensemble methods:
VEF, a novel filtering tool based on supervised learning. In particular, VEF trains a Random Forest (RF) on a variant call set from a sample for which a high-confidence set of “true” variants (i.e., a ground truth of gold standard) is available. VEF generalizes well, in that it can be trained and applied to VCF files generated from data of different coverages, as well as data produced by different sequencing machines.
□ Fast and scalable genome-wide inference of local tree topologies from large number of haplotypes based on tree consistent PBWT data structure:
an alternative form of the positional Burrows-Wheeler transform (PBWT), which they call the “tree consistent PBWT ” or shortly tcPBWT. the tcPBWT algorithm will find the correct topology of the tree in case of the perfect phylogeny (without recombinations, and with at most one mutation at each site). tcPBWT method scales linearly both in the number and in the length of sequence, and these tree topologies can capture both global population structure and local tree structure.
□ CKN-seq: Biological Sequence Modeling with Convolutional Kernel Networks:
CKN-seq is a hybrid approach between convolutional neural networks and kernel methods to model biological sequences. This method enjoys the ability of convolutional neural networks to learn data representations that are adapted to a specific task, while the kernel point of view yields algorithms.
□ Two-Tier Mapper, an unbiased topology-based clustering method for enhanced global gene expression analysis:
TTMap discerns divergent features in the control group, adjusts for them, and identifies outliers. the deviation of each test sample from the control group in a high-dimensional space is computed, and the test samples are clustered using a new Mapper-based topological algorithm at two levels: a global tier and local tiers.
□ DeepPVP: phenotype-based prioritization of causative variants using deep learning:
an extension of the PhenomeNET Variant Predictor (PVP) system which uses deep learning & achieves significantly better performance in predicting disease-associated variants than the previous PVP, as well as competing algorithms that combine pathogenicity and phenotype similarity. DeepPVP not only uses a deep artificial neural network to classify variants into causative and non-causative but also corrects for a common bias in variant prioritization methods in which gene-based features are repeated and potentially lead to overfitting.
□ scGEApp: a Matlab app for feature selection on single-cell RNA sequencing data:
This method can be applied to single-sample or two-sample scRNA-seq data, identify feature genes, e.g., those with unexpectedly high CV for given μ and rdrop of those genes, or genes with the most feature changes. Users can operate scGEApp through GUIs to use the full spectrum of functions including normalization, batch effect correction, imputation, visualization, feature selection, and downstream analyses with GSEA and GOrilla.
□ The universal decay of collective memory and attention
once we isolate the temporal dimension of the decay, the attention received by cultural products decays following a universal biexponential function. explain this universality by proposing a mathematical model based on communicative and cultural memory, which fits the data better than previously proposed log-normal and exponential models.
□ SwiftOrtho: a Fast, Memory-Efficient, Multiple Genome Orthology Classifier:
SwiftOrtho is orthology analysis tool which identifies orthologs, paralogs and co-orthologs for genomes. It is a graph-based approach. SwiftOrtho employs a seed-and-extension algorithm to find homologous gene pairs. At the extension phase, SwiftOrtho uses a variation of the Smith-Waterman algorithm, the k-banded Smith-Waterman or k-SWAT, which only allows for k gaps. k-SWAT fills a band of cells along the main diagonal of the similarity score matrix, and the complexity of k-swat is reduced to O(k · min(n, m)), where k is the maximum allowed number of gaps.
□ Adaboost-SVM-based probability algorithm for the prediction of all mature miRNA sites based on structured-sequence features:
a computational method, matFinder, that uses an AdaBoost-SVM algorithm to predict all the process sites of the mature miRNA in a pre-miRNA transcript. the AdaBoost-SVM algorithm was used to construct the classifier. The classifier training process focuses on incorrectly classified samples, and the integrated results use the common decision strategies of the weak classifier with different weights.
□ Constrained incremental tree building: new absolute fast converging phylogeny estimation methods with improved scalability and accuracy:
a new approach to large-scale phylogeny estimation that shares some of the features of DCMNJ but bypasses the use of supertree methods. this new approach is Absolute Fast Converging (AFC) and uses polynomial time and space. maximum likelihood (if solved exactly) is AFC under the standard sequence evolution models, and although it is NP-hard to solve exactly there are many seemingly good heuristics for maximum likelihood (e.g., RAxML).
□ HiGlass: web-based visual exploration and analysis of genome interaction maps:
Projects such as ENCODE and 4D Nucleome are generating Hi-C data, annotating it with metadata, and making them available to the broader public. However, there is a need to make it easier for researchers to find and integrate the data that helps answer their biological questions. HiGlass provides a rich interface for rapid, multiplex, and multiscale navigation of 2D genomic maps alongside 1D genomic tracks, allowing users to combine various data types, synchronize multiple visualization modalities, and share fully customizable views with others.
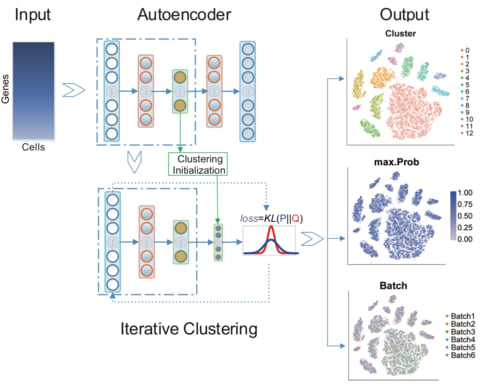
□ DESC: Deep learning enables accurate clustering and batch effect removal in single-cell RNA-seq analysis:
an unsupervised deep embedding algorithm for single-cell clustering (DESC) that iteratively learns cluster-specific gene expression signatures and cluster assignment. DESC significantly improves clustering accuracy across various datasets and is capable of removing complex batch effects while maintaining true biological variations.
□ DeepMNE-CNN: Integrating multi-network topology for gene function prediction using deep neural networks:
DeepMNE-CNN utilizes a convolutional neural network based on the integrated feature embedding to annotate unlabeled gene functions. DeepMNE-CNN mainly contains two components. One component is multi-networks integration framework, which applies a novel semi-supervised autoencoder to map input networks into a low-dimension and non-linear space based on prior information constraints. The other is CNN-based function predictor, which use convolutional neural network to learn feature embedding.
□ DeePaC: Predicting pathogenic potential of novel DNA with a universal framework for reverse-complement neural networks:
The universal framework for reverse-complement neural networks enables transformation of traditional deep learning architectures into their RC-counterparts, guaranteeing consistent predictions for any given DNA sequence, regardless of its orientation.
□ MuSiC: Bulk tissue cell type deconvolution with multi-subject single-cell expression reference:
By appropriate weighting of genes showing cross-subject and cross-cell consistency, MuSiC enables the transfer of cell type-specific gene expression information from one dataset to another. MuSiC is a weighted non-negative least squares regression (W-NNLS), which does not require pre-selected marker genes. The iterative estimation procedure automatically imposes more weight on informative genes and less weight on non-informative genes.
□ C1 REAP-seq: Fluidigm Introduces REAP-Seq for Multi-Omic Single-Cell Analysis on the C1
Information theory represents messages as spheres in high dimensional spaces. Better sphere packing leads to better communications systems. The densest known packing of hyperspheres occurs on the Leech lattice in 24 dimensions.
□ Laser light can contain intricate, beautiful fractals: Despite their simplicity, certain lasers can create the complex patterns
advance the existing theory of fractal laser modes, first by predicting three-dimensional self-similar fractal structure around the center of the magnified self-conjugate plane and second by showing, quantitatively, that intensity cross sections are most self-similar in the magnified self-conjugate plane.
□ Parametric and non-parametric gradient matching for network inference: a comparison:
In order to avoid the computational cost of large-scale simulations, a two-step Gaussian process interpolation based gradient matching approach has been proposed to solve differential equations approximately. They use model averaging, based on the Bayesian Information Criterion (BIC), to combine the different inferences. The performance of different inference approaches is evaluated using area under the precision-recall curves.
□ PROSSTT: probabilistic simulation of single-cell RNA-seq data for complex differentiation processes:
PROSSTT (PRObabilistic Simulations of ScRNA-seq Tree-like Topologies) is a package with code for the simulation of scRNAseq data for dynamic processes such as cell differentiation. PROSSTT can simulate scRNA-seq datasets for differentiation processes with lineage trees of any desired complexity, noise level, noise model, and size. PROSSTT also provides scripts to quantify the quality of predicted lineage trees.
□ Two-step graph mapper: Assessing graph-based read mappers against a novel baseline approach highlights strengths and weaknesses of the current generation of methods:
using the initial graph alignments to predict a linear path through the graph, and then re-aligning all the reads to this linear path using the linear mapper increases mapping accuracy. although the path-estimation in the first step of the two-step approach implicitly estimates variants present in the graph, the intention of this step is not to do variant calling – instead variant calling can be performed as a follow-up step based on the aligned reads.
□ Assembly Graph Browser: interactive visualization of assembly graphs:
AGB includes a number of novel functions including repeat analysis, construction of the contracted assembly graphs (i.e., the graphs obtained by collapsing a selected set of edges). AGB uses d3-graphviz, GfaPy, NetworkX-METIS, and QUAST-LG. AGB visualizes the assembly graph produced by an assembler, where edges represent various genome segments (each genome segment is represented by its forward and reverse-complement edge).
□ Network hubs affect evolvability: how alterations in a gene central to a network affect evolutionary processes:
Fitness landscape and possible evolutionary trajectories. Perturbing a hub gene or a peripheral gene can both lead to a decrease in fitness, but the number of available evolutionary trajectories is higher when a hub gene is perturbed. adaptation to an altered hub occurred by optimizing the subnetworks the hub is connected to and not by restoring the hub itself. These subnetworks were different between the populations, and as a result, the evolved lineages showed a large variety in their phenotypic profiles.
a method to attribute the cause of anomalies to either the incompleteness of the reference transcriptome or the algorithmic mistakes, and this method precisely detects misquantifications with both causes. Applying anomaly detection to 30 GEUVADIS and 16 Human Body Map samples, they detect 103 genes with potential unannotated isoforms.
□ Performance of neural network basecalling tools for Oxford Nanopore sequencing:
Albacore, Guppy and Scrappie all use an architecture that ONT calls RGRGR – named after its alternating reverse-GRU and GRU layers. To test whether more complex networks perform better, they modified ONT’s RGRGR network by widening the convolutional layer and doubling the hidden layer size.
□ Waddington-OT: Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming:
Waddington-OT, an approach for studying developmental time courses to infer ancestor-descendant fates and model the regulatory programs that underlie them. applying the method to reconstruct the landscape of reprogramming from 315,000 single-cell RNA sequencing (scRNA-seq) profiles, collected at half-day intervals across 18 days.
□ Coordinate-based mapping of tabular data enables fast and scalable queries:
Across the subfields of biology, researchers store a considerable proportion of tabular data in plain-text formats. This approach coincides with the Unix and “Pragmatic Programming” philosophies, which advocate for storing data and sharing data among computer programs as plain text.
The HDF5 format is designed primarily for numerical data, whereas we sought the ability to handle other data types as well. As a columnar storage solution, Parquet was efficient at projection.
□ Formal axioms in biomedical ontologies improve analysis and interpretation of associated data:
The axioms and meta-data of different ontologies contribute in varying degrees to improving data analysis. the formal axioms that have been created for the Gene Ontology and several other ontologies significantly improve ontology-based prediction models through provision of domain-specific background knowledge.
□ Biomedical Concept Recognition Using Deep Neural Sequence Models:
Deep learning methods for span detection were equivalent in performance to traditional conditional random field methods. As natural training data is limited to the concepts used in CRAFT annotations, the addition of synthetic training data, class names and synonyms, to the normalization step has the potential to improve recall on class not in CRAFT. The CRF+OpenNMT system also outperforms the other systems for most ontologies and is the best-performing system for the GO_BP/MF annotation set.
□ ComPath: an ecosystem for exploring, analyzing, and curating mappings across pathway databases:
Although the absence of topological pathway information in ComPath is an irrefutable limitation in this study, gene-centric approaches enable a reduction of complexity in pathway comparison as well as integration of resources which do not provide topology information.
□ ChimeraUGEM: unsupervised gene expression modeling in any given organism:
ChimeraUGEM provides tools for the analysis of gene sequences (coding and non-coding), as well as the design of protein coding sequences for optimized expression, based on the Chimera algorithms and codon usage optimization.
□ netNMF-sc: Leveraging gene-gene interactions for imputation and dimensionality reduction in single-cell expression analysis:
netNMF-sc combines network-regularized non-negative matrix factoriza- tion with a procedure for handling zero inflation in transcript count matrices. The matrix factorization results in a low-dimensional representation of the transcript count matrix, which imputes gene abundance for both zero and non-zero entries and can be used to cluster cells.
□ Spinning convincing stories for both true and false association signals
What are some of the additional applications of Practical Byzantine Fault Tolerance outside of the most obvious use in blockchain?
There're many challenges with their practical implementation in real-world systems, but there're many promising scenarios where it'd be extremely beneficial, starting with flight control/spacecraft flight systems, and other systems that need agreement and expect Byzantine errors.
□ Mesh: Compacting Memory Management for C/C++ Applications
Mesh combines novel randomized algorithms with widely-supported virtual memory operations to provably reduce fragmentation, breaking the classical Robson bounds with high probability.
□ Transcript expression-aware annotation improves rare variant discovery and interpretation
In gnomAD we see variants we don't expect (e.g. in haploinsufficient disease genes). Often found on alternative txs, with little evidence of expression. he pext score, which summarizes isoform expression for variants. Regions with high pext are more conserved, and nonsynonymous variation on them is more deleterious. Opposite true for low pext regions, which are enriched for false exon annotations.
Mathematical models of biology can predict the relative contribution of a gene to a specific function of a pathway. The method combines gene weights from model predictions and gene ranks from genome-wide association studies into a weighted gene-set test.
□ clonealign assigns single-cell RNA-seq expression to clones by probabilistically mapping RNA-seq to clone-specific copy number profiles using reparametrization gradient variational inference:
Metacosmos is constructed around the natural balance between beauty and chaos – how elements can come together in (seemingly) utter chaos to create a unified, structured whole.
The idea and inspiration behind the piece, which is connected as much to the human experience as to the universe, is the speculative metaphor of falling into a black hole – the unknown – with endless constellations and layers of opposing forces connecting and communicating with each other, expanding and contracting, projecting a struggle for power as the different sources pull on you and you realize that you are being drawn into a force that is beyond your control.
Berliner Philharmoniker appのDigital Concert Hallにて、Alan Gilbert指揮のベルリン・フィルハーモニー管弦楽団の公演を視聴。
アイスランドの作曲家、Anna Thorvaldsdottirの《Metacosmos (メタコスモス)》はヨーロッパ初演。
宇宙的なスケールの鳴動を思わせる荘厳な響き.
同時上演のSergei Prokofiev "Concerto for Violin and Orchestra No. 2 in G minor, op. 63"ではLisa Batiashviliとの競演。
































































{kind=link}