









□ "Lesson learned. Lesson ignored. My soul has charred, what they burned."
黒々とした支脈が擬ひの灯火を垂れて、燦爛と赫くベツレヘムの星を穿ってゐる。この樫を砥いで己が諱を削り出そう。何人も目もくれぬ此の欺瞞の陰の縁に、やがてなる虚妄の姿を窶す為に。其れを希う心は疵となり、塞ぐ眼は真珠となる。誰も耳を敧てぬ、空より高くいやはてに刻んだ、愛、愛、愛の名前を
□ 「形だけが遺る」ことは、形こそが重要なのであり、それ自体が力学の意味を映している。
□ 世界の表面はジッパーのようなもので、前へ閉じても、後ろへ開いても、その境界線上の型は噛み合わさっている。可逆・不可逆は時間の表現ではなく、まさにその型の連なりを示している。
□ 神や運命、或いはそれらに準ずる法則めいた何かが在るかどうか、または信じる、信じないの問いかけすら実に狭隘であるとしか言いようがないのだけど、不可知論の立場を取るまでもなく、それらは等しく可能世界の無限に近い階層の、どこで干渉を生じるかという論点を未定義に置き換えたものに過ぎない。
□ 生き方にしろなんにしろ、誰かが口出しようとしまいと、変わるものは変えようとしなくても変わる。安らぎや幸せなんてものは通過点でいい。苛烈に生きたいと欲したのは自分自身だ。口を衝いて語り出でるものの舌触りだけで、確からしいと感じていた。この径の先を行くだけでは辿り着けないのに。

□ Novel silicon phases and nanostructures for solar energy conversion
>> http://ow.ly/rImq306TLw9
□ Deep Learning for RegEx:
>> http://dlacombejr.github.io/2016/11/13/deep-learning-for-regex.html
Extract basic features from input using 1D convolutions of varying lengths (1, 3, 5, 7 with 75, 75, 50, and 50 fiters for each length.) fed through a series of blocks that perform 1D à trous convolutions w/ Batch Normalization, Rectified Linear Units & skip connections. This allowed the network to choose the best matching start and end indices based on scopes that covered almost the entire input due to the dilated convolutions. two 1D convolutional filters with softmax activations were performed over the residual output; the maximum argument represented the index of highest probability for the start and end indices.
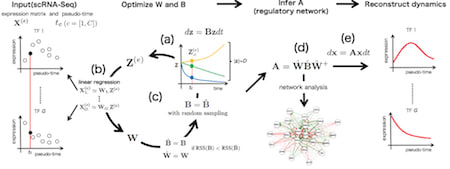
□ SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation:
>> http://biorxiv.org/content/biorxiv/early/2016/11/21/088856.full.pdf
SCODE is based on the transformation of linear ODEs and linear regression, and the time complexity is significantly small. The advancement of scRNA-Seq and the analysis of differentiation reconstruction and pseudo-time have elucidated differentiation mechanisms.
pseudotime <- read.table(ftime, sep="\t")[1:cnum,2]
pseudotime <- pseudotime/max(pseudotime)
sample_Z <- function(){
for(i in 1:pnum){
for(j in 1:cnum){
Z[i,j] <<- exp(new_B[i]*pseudotime[j]) + runif(1, min=-0.001, max=0.001)
□ HiC-Spector: A matrix library for spectral and reproducibility analysis of Hi-C contact maps:
>> http://biorxiv.org/content/biorxiv/early/2016/11/21/088922.full.pdf
a chromosomal contact map by a symmetric and non-negative adjacency matrix 𝑊. The matrix elements represent the frequencies of contact between genomic loci and therefore serve as a proxy of spatial distance. The normalized Laplacian matrix is closely related to random walk processes taking place in the underlying graph of 𝑊.
the smallest ev of L is 0. in many networks, because of the existenc of singleton, we expect more than 1 zero ev. if we do normalization, 0=lambda1<=lambda_1<=lambda_2.,,,<=2
to avoid matrix multiplication with 0, inf. actually
L_norm(i,j)=1 if i=j
L_norm(i,j)=-1/sqrt(deg(i)*deg(j)))
0 otherwise.

□ Improved Measures of Integrated Information:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005123
a simple taxonomy of Φ-measures where they are each characterized by their choice of factorization method, choice of probability distributions to compare and choice of measure for comparing probability distributions. graph connectivity can be determined in polynomial time, enabling to rapidly find a good approximation for the “cruelest cut” bipartition. the time-evolution of the system state as a Markov process defined by a transition matrix M: when the system state evolves from x0 to x1, the corresponding probability distribution evolves from p0 to p1 ≡ Mp0.

□ Interpreting Finite Automata for Sequential Data:
>> https://arxiv.org/abs/1611.07100v1
As probabilistic deterministic automata models are almost as expressive as HMMs. the RTI+ state-merging algorithm for timed automata, analyze car following behavior of human drivers. In wind speed prediction, discover structure in the data by using by inferring transition guards over a potentially infinite alphabet, effectively discovering a clustering as transition labels from the sequences automatically.
□ Genomic Data Retrieval with R:
>> https://github.com/HajkD/biomartr
For automating the retrieval process on a meta-genomic scale, biomartr provides useful interface functions for genomic sequence retrieval.
Combine Evolutionary Transcriptomics with `biomartr` %\VignetteIndexEntry{Evolutionary Transcriptomics using biomartr} %\VignetteEngine{knitr::rmarkdown} %\usepackage[utf8]{inputenc}
□ @nanopore Rapid sequencing kit v2 now available at the ONT store:
https://store.nanoporetech.com/rapid-sequencing-kit-2.html
Designed for the newest R9.4 flow cells, 450nt/s
□ Knowledge Formalization and High-Throughput Data Visualization Using Signaling Network Maps:
>> http://biorxiv.org/content/biorxiv/early/2016/11/24/089409.full.pdf
a workflow for construction and annotation of signaling maps in CellDesigner, preparing the hierarchical modular structure of maps and also generation of different levels of the maps view, to allow semantic zooming-based exploration of maps in NaviCell.
□ Piecewise Deterministic Markov Processes for Continuous-Time Monte Carlo:
>> https://arxiv.org/pdf/1611.07873v1.pdf
the continuous-time sequential Monte Carlo methods seem particularly well-suited for implementation on a distributed computing architecture, as evolution of particles can be carried out in parallel.
λ(x,v)−λ(x,−v) = E(max{0,v·U(x)})−E(max{0,−v·U(x)}) = E(max{0,v·U(x)}−max{0,−v·U(x)})
= E(max{0,v·U(x)}+min{0,v·U(x)})=E(v·U(x)).
□ Graph regularized, semi-supervised learning improves annotation of de novo transcriptomes:
>> http://biorxiv.org/content/biorxiv/early/2016/11/25/089417.full.pdf
GRASS is best considered as a method capable of “boosting” an initial set of annotations by accounting for expression and sequence similarities within the de novo assembly. Quantification results obtained using Sailfish or Salmon which are used to improve the graph & filter out spurious contigs from the assembly
□ Inference of evolutionary jumps in large phylogenies using L ́evy processes:
>> http://biorxiv.org/content/biorxiv/early/2016/11/26/089276.full.pdf
an MCMC algorithm in which we can update the inverse of the above mentioned variance-covariance matrix directly without inversion when sampling jump configurations with fixed hierarchical parameters. the location of jumps can then be inferred under an empirical Bayes framework in which the hierarchical parameters are fixed to ML estimate and the developed MCMC algorithm is used to obtain for each branch the posterior probability that a jump occurred at this location.
□ Nanocall: an open source basecaller for Oxford Nanopore sequencing data:
>> http://bioinformatics.oxfordjournals.org/content/early/2016/10/07/bioinformatics.btw569.full
Nanocall produces reads comparable in mappability and quality to Metrichor 1D reads with ∼68% identity. As an important technical difference from Metrichor, with Nanocall that double-strand pore model scaling seems to work better than single-strand scaling, suggesting that the latter might lead to model overfitting. Metrichor on R9 uses a more elaborate RNN-based approach, compared to the simple HMM-based one in Nanocall.
□ LeARN: a platform for detecting, clustering and annotating non-coding RNAs:
>> http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-21
It integrates tools and web interfaces covering the three layers of the ncRNA annotation process, a flexible detection and clustering pipeline, a RNAML database and a web interface to manage the expert annotation.
□ BLISS: quantitative and versatile genome-wide profiling of DNA breaks in situ:
>> http://biorxiv.org/content/biorxiv/early/2016/12/04/091629.full.pdf
robust quantification by using UMIs to reduce data noise and count DSBs that form in different cells at the same genomic location. ~12 active work-hours over 5 days to process 24 samples by BLISS versus at least ~60 active work-hours over 15 days by BLESS.
□ Einstein Epigenomics @EpgntxEinstein
"The lesson that functional genomic assays are influenced by underlying cell subtype heterogeneity. Often ignored in expression studies."
>> https://twitter.com/EpgntxEinstein/status/807247059615121408
□ Tuuli Lappalainen @tuuliel
".@EpgntxEinstein Genetic effects are way less tissue-spec than expr/meth/etc, and ignoring cell types leads to false negs, not false pos."

□ Cox-nnet: an artificial neural network method for prognosis prediction on high-throughput omics data:
>> http://biorxiv.org/content/biorxiv/early/2016/12/11/093021.full.pdf
Some features of Cox-nnet include parallelization and GPU usage for high computational efficiency, training optimization methods such as the Nesterov accelerated gradient and flexibility in allowing the specification of neural network architecture. Incorporation of biological information into the structure or using a deep learning approach are possible.
In over 10 TCGA RNA-Seq data sets, Cox-nnet achieves a statistically significant increase in predictive accuracy, compared to the other three methods including Cox-proportional hazards (Cox-PH), Random Forests Survival and CoxBoost. The outputs from the hidden layer node can provide a new approach for survival-sensitive dimension reduction.
In Theano, the partial log likelihood is:
pl=T.sum((theta - T.log(T.sum(T.exp(theta) * R,axis=1))) * C)
□ LSTMを超える期待の新星、QRNN
>> http://qiita.com/icoxfog417/items/d77912e10a7c60ae680e
QRNN、QUASI-RECURRENT NEURAL NETWORKSとは、RNNの機構をCNNで「疑似的(QUASI)に」実装するというモデルです。
>> http://metamind.io/research/new-neural-network-building-block-allows-faster-and-more-accurate-text-understanding/
Despite lacking trainable recurrent layers, stacked QRNNs have better predictive accuracy than stacked LSTMs of the same hidden size. Due to their increased parallelism, they are up to 16 times faster at train and test time.
□ Random versus maximum entropy models of neural population activity:
>> http://biorxiv.org/content/biorxiv/early/2016/12/09/092973.full.pdf
Although the motivations of maximum entropy seem intuitive and can be formalized rigorously, the perceived arbitrariness of its assumptions has led to question its validity. Maximum entropy’s crucial - and arguably debatable - assumption is to pick, out of the many models that satisfy that constraint, the one with the largest Gibbs entropy.
□ Historian: accurate reconstruction of ancestral sequences and evolutionary rates:
>> http://biorxiv.org/content/biorxiv/early/2016/12/11/093161.full.pdf
Historian combines an efficient reimplementation of the ProtPal algorithm with performance-improving heuristics from other alignment tools. The reconstructed parent profile is a weighted finite-state transducer sampled from the posterior distribution implied by the children. The posterior probability threshold for inclusion in the parent profile and max number of states in the parent profile can both be tweaked to trade off sensitivity vs performance.
□ DeepVariant: Creating a universal SNP and small indel variant caller with deep neural networks:
>> http://biorxiv.org/content/biorxiv/early/2016/12/14/092890.full.pdf
DeepVariant can learn to call variants in a variety of sequencing technologies from deep whole genomes from 10X Genomics to Ion Ampliseq. The accuracy of DeepVariant will likely improve by transitioning to more expressive tensor-based models specialized for genomic data.
□ HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies:
>> http://genome.cshlp.org/content/early/2016/12/09/gr.213462.116
Using HapCUT2, haplotype assembly from a 90x coverage whole-genome Hi-C dataset yielded high-resolution haplotypes, 78.6% of variants phased in a single block with high pairwise phasing accuracy (~98% across chromosomes).
□ Anti-entropic virucidal energy as information:
>> http://rna-mediated.com/anti-entropic-virucidal-energy-as-information/
traveling waves can adiabatically deform as the system is increased in size without the increase in node number that would be expected for an oscillatory version of a Turing instability containing an allowed wavenumber band with a finite minimum.
□ Helical reconstruction in RELION:
>> http://biorxiv.org/content/biorxiv/early/2016/12/17/095034.full.pdf
a new implementation for the reconstruction of helical assemblies in the empirical Bayesian framework of RELION. This approach calculates optimal linear filters for the 3D reconstruction by embedding helical symmetry operators in Fourier-space, and deals with deviations from perfect helical symmetry through Gaussian-shaped priors on the orientations of individual segments.
□ FlashPCA2: principal component analysis of biobank-scale genotype datasets:
>> http://biorxiv.org/content/biorxiv/early/2016/12/17/094714.full.pdf
FlashPCA2, which outperforms existing tools in terms of computation time on large datasets (n = 1,000,000 individuals and 100,000 SNPs), while utilising bounded memory and maintaining high accuracy for the top eigenvalues/eigenvectors.
□ DeepBach: a Steerable Model for Bach chorales generation:
>> https://arxiv.org/pdf/1612.01010v1.pdf
□ DeepBach: harmonization in the style of Bach generated using deep learning
DeepBach using Keras with the Tensorflow backend. DeepBach is able to produce coherent musical phrases and provides, for instance, varied reharmonizations of melodies without plagiarism.

□ GeNNet: An Integrated Platform for Unifying Workflow Management and Graph Databases for Transcriptome Data Analysis:
>> http://biorxiv.org/content/biorxiv/early/2016/12/18/095257.full.pdf
GeNNet database (GeNNet-DB) schema is based on the Neo4j database management system. In the GeNNet platform there is an initial database defined by interactions between genes as described in Background preparation.
MATCH (e:EXPERIMENT)-[s:Was_selected]-> (g:GENE)-[p:PPI_interaction]-(h:GENE)- [:Was_clusterized]-(c:CLUSTER)- [:Was_represented]-(b:BP)
WHERE p.combined_score > 800
WITH DISTINCT g, COUNT(distinct h) AS score
WHERE score > 50 WITH collect(g) AS gs CREATE (hub:Hub {name: ’HUB’})
WITH gs, hub UNWIND gs AS g
CREATE (g)-[:AS_HUBS]->(hub)
RETURN *
□ Partitioned learning of deep Boltzmann machines for SNP data:
>> http://biorxiv.org/content/biorxiv/early/2016/12/20/095638.full.pdf
For subsequently identifying relevant hidden units and extracting SNPs, univariate Cox regression models were used, with additive (0/1/2) coding to avoid convergence issues. When fitting Cox models for each of the original 70 SNPs, no SNP was found to be significant after Bonferroni-Holm correction (FWER ≤ 0.05).
□ Integrated deep learned transcriptomic and structure-based predictor of clinical trials outcomes:
>> http://biorxiv.org/content/biorxiv/early/2016/12/20/095653.full.pdf
Remarkably, the results of iPANDA-based DNNs for side effects significantly outperform gene-based DNNs. The search space for all DNNs was: from 100 to 1000 neurons, from 3 to 6 layers, Adam, Adadelta, Adagrad as optimizers, ReLU, PReLU, ELU as activation functions, from 0.25 to 0.5 as dropout probability.
All of the authors are employed by Insilico Medicine, Inc at the Pharmaceutical Artificial Intelligence (Pharma.AI) division.
□ Symmetry of Lyapunov exponents in bifurcation structures of one-dimensional maps:
>> http://aip.scitation.org/doi/full/10.1063/1.4972401
the symmetry in the LE emerges for three maps—the logistic map, the chaotic neuron model, and the odd-logistic map this symmetry might be observed in other types of dynamical systems.
The logistic map, x↦μx(1−x),
The chaotic neuron model, y↦0.7y−1/(1+e−y/0.02)+b
The odd-logistic map, z↦γz(1−z2)
□ Beyond the Hypercube: Evolutionary Accessibility of Fitness Landscapes with Realistic Mutational Networks:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005218
the accessibility of the fittest organism depends on the number of types of basic “units” used to encode genotypes. Equivalently to re-indexing sub-landscapes of the real fitness landscape one could adapt the DFS algorithm and related structures storing accessible pathways to have arbitrary target and initial genotypes, not encoded by sequences {K − 1, K − 1,…, K − 1} and {0, 0, …0}.