
□ Limacina antarctica (Swimming sea snail) by Alexander Semenov.
"The phenomena resulting from the existence of one or more engrams in an organism, I describe as mnemic phenomena. The totality of the mnemic potentialities of an organism is its "Mneme""
- Richard Wolfgang Semon.
"Lang ist Die Zeit, es ereignet sich aber Das Wahre."
-Hölderlin: Mnemosyne.
□ Quantum diffusion with drift and the Einstein relation.: The ergodic average over time of the state of the particle
>>
http://scitation.aip.org/content/aip/journal/jmp/55/7/10.1063/1.4881532 …
a quantum particle dynamics driven by a constant external force & a non-equilibrium steady state describing a non-zero mean drift particle.
anti-linear time-reversal operator Θ = Θ S ⊗ Θ R, where Θ S is given by
ΘSf(x)=f(x), f∈ℓ^2(Λ),

□
mason_lab:
The PromethION is live at NYGC!
□
iNut:
nmoporeの現時点での実用性はともかく、この小さいモジュールを大量に並列させることで自在にスケールできるというのはデザインとしてとてもよいとおもう、MapReduceとかクラウドインフラのオートスケールなんかに通じるものがある、頑張ってイルミナ一強時代を打破してほしい

□ BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers:
http://nar.oxfordjournals.org/content/early/2014/11/26/nar.gku1211.full …
BreaKmer had a 97.4% overall sensitivity for known events and predicted 17 positively validated, novel variants.

□ MGAS: a powerful tool for multivariate gene-based genome-wide association analysis compared to GATES and MANOVA:
>>
http://bioinformatics.oxfordjournals.org/content/early/2014/11/25/bioinformatics.btu783.full.pdf
Schematic representation of six trait-generating genotype- phenotype models. (f) all phenotypes are bi-directionally related, w/ relations being stronger, variance-covariance matrix mimicking that of a 4-factor model.

An alignment of three genomes with eleven blocks in all four graph representations.
□ Genome alignment with graph data structures: a comparison: comparison of de Bruijn / Enredo / cactus graph approach
>>
http://www.biomedcentral.com/1471-2105/15/99
Considering the large search space, genome alignment is an ambitious task and is usually accomplished using heuristic approaches. The first step in genome alignment is commonly the computation of a set of local alignments.

these quantities give rise to the OBC, and MMSE and MSE estimates for the error as described in the text.
□ MCMC implementation of the optimal Bayesian classifier for non-Gaussian models: model-based RNA-Seq classification:
>>
http://www.biomedcentral.com/content/pdf/s12859-014-0401-3.pdf …
非ガウスモデルを用いた配列データ分類のための階層的多変量ポアゾンモデルおよび最適ベイズ識別: 合成データセットに関するベイズ最小二乗誤差(MMSE)の分類を特徴空間上で計算し実証する。
a one-dimensional simplification of the MP model
Var(X) = E[Var(X|λ)] + Var(E[X|λ])
= e(μ+σ2/2) + (eσ2 - 1)e(2μ+σ2) ≥ eμ = Var(Poisson(eμ))
MP OBC setting for synthetic data
μy ∼Normal(0, 0.2),
σy ∼Inverse-Gamma(1, 3),
ρ = Uniform(0.0, 1.0),
dlow = 9,
dhigh = 11,
blocksize = 5.
The resulting provides superior classification performance compared to state-of-the-art classifiers such as SVM with a radial basis kernel.
Owing to limited available computational resources, we could only allocate around 30 seconds on a single CPU core for each MCMC run. This necessitated using only four genes for these classification results as each iteration of MCMC procedure has time complexity of O(D^3)
□ Zélus : A Synchronous Language with ODEs
>>
http://lambda-the-ultimate.org/node/5087
Zélus is a new programming language for modeling systems that mix discrete logical time and continuous time behaviors. Combinatorial values are imported from Objective Caml and programs are compiled into Objective Caml code.
The standard way to detect events in a numeric solver is via zero-crossings where a solver monitors expressions for changes in sign and then, if they are detected, searches for a more precise instant of crossing.
SOAS with lead network
s = poly(0, 's')
[a, b, c, d] = abcd(tf2ss(1.2 * (s + 5) / (s + 15)))
□ ‘N-of-1-pathways’ unveils personal deregulated mechanisms from a single pair of RNA-Seq: towards precision medicine
>>
http://jamia.oxfordjournals.org/content/21/6/1015 …
Nof1 is not novel, as this has been done using meta-analyses or Bayesian-mixed model for generalizing the value of therapeutic interventions.
□ Rapid evaluation and quality control of next generation sequencing data with FaQCs
http://bit.ly/1tYGKOT
Fold Coverage =( peak k‐mer coverage)×L/(L-K+1)
□ Wardrobe Experiment Management System for Integrated Analysis of Epigenomics Data:
http://biorxiv.org/content/early/2014/12/18/012799 …
Predefined pipelines allow users to download data from core facilities or public databases, calculate RPKMs and identify islands. Advanced capabilities include differential gene expression and binding analysis, and creation of average tag density profiles and heat maps. Wardrobe uses the MAnorm algorithm, which produces fold changes & p-values for the peaks, the areas of interest can be viewed on the browser
□ PANDORA: Systematic integration of RNA-Seq statistical algorithms for accurate detection of DGE patterns
>>
http://nar.oxfordjournals.org/content/early/2014/12/01/nar.gku1273.long …
metaseqR can combine the P-value scores returned by the application of more than one statistical tests with six approaches.
□ Ulysses: accurate detection of low-frequency structural variations in large insert-size sequencing libraries:
>>
http://bioinformatics.oxfordjournals.org/content/early/2014/11/27/bioinformatics.btu730.short …
Ulysses achieves high specificity over the complete spectrum of variants by assessing, in a principled manner, the statistical significance of each possible variant against an explicit model for the generation of experimental noise.
□ VSEARCH: Open 64-bit multithreaded tool for processing metagenomic sequences, clustering, chimera detection, etc…
>>
https://github.com/torognes/vsearch …
VSEARCH stands for vectorized search, as the tool takes advantage of parallelism in the form of SIMD vectorization as well as multiple threads to perform accurate alignments at high speed. VSEARCH uses an optimal global aligner (full dynamic programming Needleman-Wunsch), in contrast to USEARCH which by default uses a heuristic seed and extend aligner.

□ Alignment by the numbers: sequence assembly using reduced dimensionality numerical representations:
>>
http://biorxiv.org/content/biorxiv/early/2014/11/28/011940.1.full.pdf …
a read graph with weighted edges, the shortest path in the graph is identified with a breadth-first search algorithm
Suitable methods for measuring the similarity of sequential data include the Lp-norms, Dynamic Time Warping, Longest Common SubSequence, and alignment algorithms such as the Needleman-Wunsch and Smith-Waterman algorithms.
i) transforming symbolic nucleotide sequences to numerical sequences,
ii) creating approximate representations of reads and, where appropriate, of a reference sequence
iii) performing accelerated comparison of the reduced dimensionality sequence representations to identify candidate alignments
iv) verifying and finishing candidate alignments using original, full resolution sequences.
□ DEEP: a general computational framework for predicting enhancers:
>>
http://nar.oxfordjournals.org/content/early/2014/11/05/nar.gku1058.long …
results on (FANTOM5 DEEP-FANTOM) data where DEEP achieves 90.2% accuracy and 90% geometric mean (GM) of specificity and sensitivity.
a. Positive Predictive Value (PPV) = A/B
b. Jaccard Index = A/(B + C - A)
c. F1-score = 2*A/(B + C)
d. Promoter overlap fraction (POF) = D/B
□ Canadian BiogeniQ to challenge @23andMe in direct-to-consumer genetic testing domain
http://business.financialpost.com/2014/11/30/why-biogeniq-may-have-an-edge-on-google-backed-23andme … #genomics HT @drbachinsky
□ A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping:
http://www.cell.com/cell/abstract/S0092-8674%2814%2901497-4 …
□ UCLA creates new Institute for Quantitative and Computational Biosciences focused on tools for personalized medicine.
http://bit.ly/13lq3qI
□ 東芝、ゲノム解析に参入 15年度売上高30億円目標 - SankeiBiz
http://bit.ly/1yb35QP
□
hinaichigo:
東芝ヘルスケアはaffymetrixのAxiom でGenotype研究やるみたいね。
http://www.businesswire.jp/news/jp/20141130005021/ja …
□ Global Next Generation Sequencing Market to grow at a CAGR of more than 20% to reach around $5.0 billion by 2020:
http://www.researchandmarkets.com/research/g47zz8/global_next …

(An asynchronous Boolean multiplex control network. )
□ Controllability of asynchronous Boolean multiplex control networks:
>>
http://scitation.aip.org/content/aip/journal/chaos/24/3/10.1063/1.4887278 …
calculate the control-depending network transition matrix of system. at time t, x˜i(t)=xli(t), l∈{1,2,…,k˜} . when node 1.
x˜(t+1)=McMnx˜13(t)u1(t)x˜2(t)x˜3(t)x˜4(t)=McMn(I2⊗W[8,2])W[2](I2⊗Mr)W[2,8]EdW[2]u1(t)x˜1(t)x˜2(t)x˜3(t)x˜4(t)≜ Lˆ1u(t)x˜(t).
The onset of M(mitosis) & S(DNA replication) phases of the cell cycle are controlled by the periodic activation of cyclin-dependent kinases. the differential equations model of the above process and its corresponding Boolean model were proposed by Romond and Heidel respectively.
□ SnakeViz: Visually profile your Python code with D3.js: a graphical viewer for the output of Python’s cProfile module
>>
http://jiffyclub.github.io/snakeviz/
var partition = d3.layout.partition()
.size([2 * Math.PI, radius * radius])
.value(function(d) { return d.size; });
var y = d3.scale.linear().domain([0, radius * radius]).range([0, radius]);
var arc = d3.svg.arc()
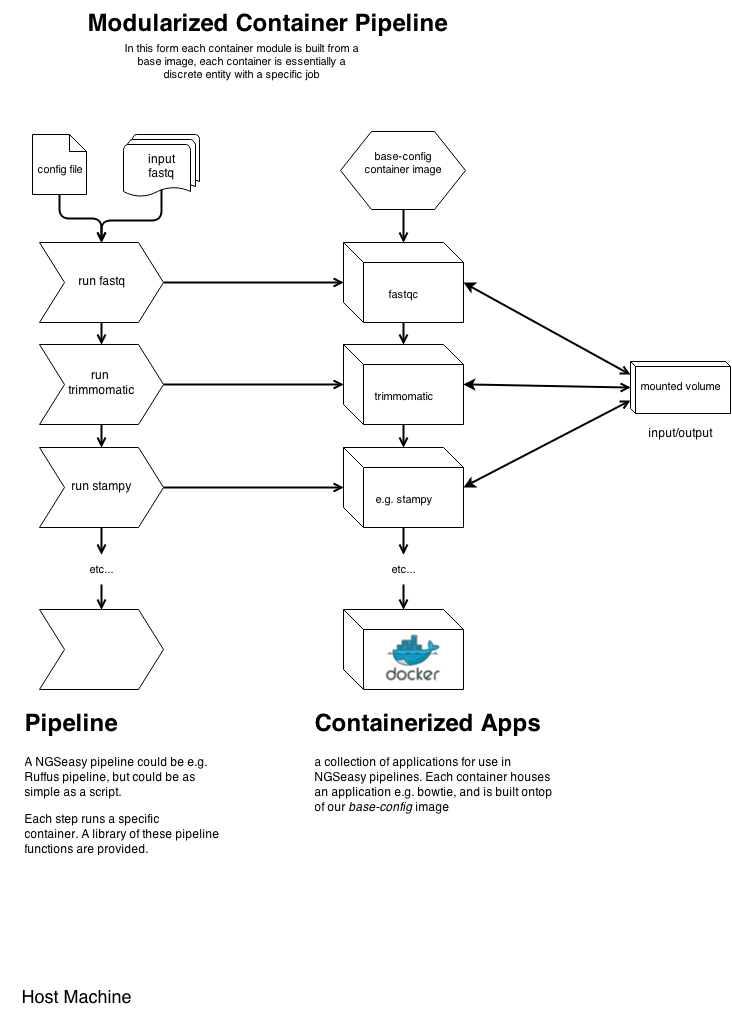
□ NGSeasy: A Dockerized Next Generation Sequencing Pipeline (QC, Align, Calling, Annotation): 12 Steps to NGS Freedom
>>
http://khp-informatics.github.io/ngs/
NGSeasy Pipeline Components: QC of raw fastq files, SAM/BAM manipulation, alignment, first pass variant discovery.
NGSeasy allows users with minimal computational/bioinformatic skills to set up an NGS analysis on their own samples on any infrastructure.

Integrative analysis of public ChIP-seq experiments reveals a complex multi-cell regulatory landscape:
>>
http://nar.oxfordjournals.org/content/early/2014/12/03/nar.gku1280.abstract …
Network representations of TFs co-localization across the genome. Highlighted subnetworks of highly connected and strongly specific TFs with functional annotations. Barplots represent Gene Ontology Biological Process enrichments calculated by DAVID (x-axis = -log10 Benjamini score).
□ Frameshift alignment: statistics and post-genomic applications:
>>
http://bioinformatics.oxfordjournals.org/content/30/24/3575.short …
□ VTBuilder: a tool for the assembly of multi isoform transcriptomes:
http://www.biomedcentral.com/content/pdf/s12859-014-0389-8.pdf …
□ Beaker: notebook development environment w/ complex datasets: Python, R, Julia, Groovy, JavaScript, Node, Ruby, LaTeX
http://beakernotebook.com
□
chrismentzel
for me,
#DDDi == Data-Driven Discovery Investigators:
http://bit.ly/DDDinvestigators … and #DDD == the initiative itself:
http://bit.ly/MooreData
□ Fourier transform infrared spectroscopy of ligand photodissociation and migration in inducible nitric oxide synthase:
http://f1000research.com/articles/3-290/v2 …
□
_masaka:
BabelNet 3.0 now out http://lists.w3.org/Archives/Public/semantic-web/2014Dec/0079.html … WordNet、Wikipedia、Wiktionaryなどのより高精度な統合、271言語、1370万意味単位、20億トリプル。新APIが使えるベータプログラム1/31まで実施

□ The lunar dynamo: ancient core dynamo field persisted from at least 4.25-3.56 Bn years ago
>>
http://www.sciencemag.org/content/346/6214/1246753.abstract …
The extended history of the lunar dynamo appears to demand long-lived power sources such as mantle precession and core crystallization.
□ A geometric approach to the Landauer-Büttiker formula:
>>
http://scitation.aip.org/content/aip/journal/jmp/55/7/10.1063/1.4879238 …
n ideal Fermi gas confined to a geometric structure consisting of a central region – the sample – connected to several infinitely extended ends―the reservoirs. Under physically reasonable assumptions on the propagation properties of the one-particle dynamics within these reservoirs, we show that the state of the Fermi gas relaxes to a steady state.
Ruelle's scattering approach also works in the presence of weak local interactions (many body interactions are sufficiently well localized) In this case, Møller operator of Hilbert space scattering theory is replaced by a Møller morphism acting on the C*-algebra 𝒪 of observables.
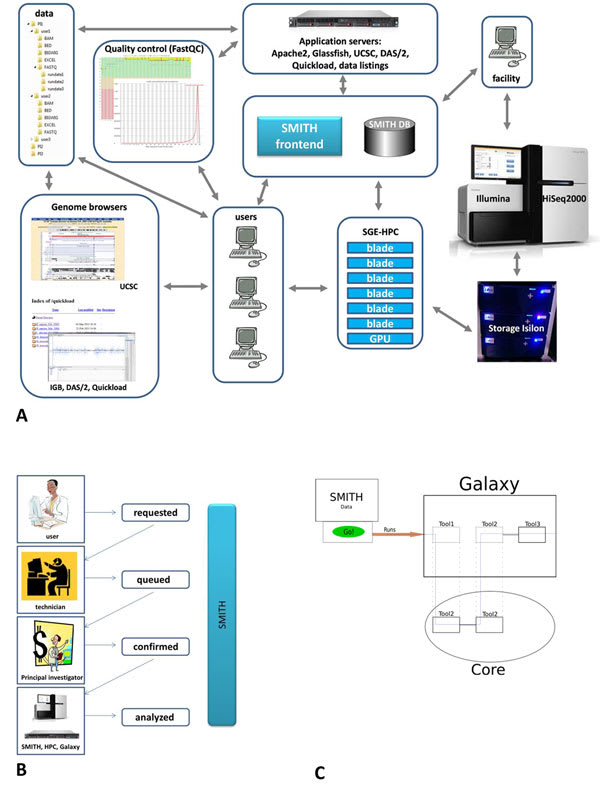
□ SMITH: a LIMS for handling next-generation sequencing workflows
>>
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4255740/ …
SMITH Infrastructure: Sequencing is performed on Illumina HiSeq2000. Isilon mass storage & Sun Grid Engine cluster.
SMITH architecture is divided into a web-tier, a middle-tier, an information system tier, adheres to the Model-View-Controller(MVC) paradigm.
Genologics Clarity LIMS is a highly customisable LIMS with a rich set of options and short deployment time.

□ Signaling complexes are unexpectedly flexible; good news for evolution & #SynBio #PLOSBiology
>>
http://plos.io/1z6zINp
□ Oscillation-Induced Signal Transmission and Gating in Neural Circuits | @PLoSCompBiol
http://ow.ly/FLWVH
□ New avian tree of life charts how #birds evolved from dinos:
http://n.pr/1zUiAdn RT@gbrumfiel w/@erichjarvis
□ Avian phylogenomics meets Jurassic Park: reconstructing a dinosaur genome (structure)
http://www.biomedcentral.com/1471-2164/15/1060 … #noamber
□ Core and region-enriched networks of behaviorally regulated genes and the singing genome:
>>
http://www.sciencemag.org/content/346/6215/1256780.full …
10% of the genes in the avian genome were regulated by singing, and found a striking regional diversity of both basal and singing-induced programs in the four key song nuclei of the zebra finch, a vocal learning songbird. epigenetic mechanism is just beginning to be explored at the level of neural activity and has not been addressed in complex behaviors.
□ MusicMood - Machine Learning in Automatic Music Mood Prediction Based on Song Lyrics:
>>
http://www.slideshare.net/SebastianRaschka/musicmood-20140912 …
the class-conditional probabilities of the a multinomial naive Bayes model are calculated as
P̂ (xi∣ωj)=[∑tf(xi,d∈ωj)+α]/[∑Nd∈ωj+α⋅n]
the class-conditional probability of encountering the lyrics x is calculated as the product of the likelihoods of the individual terms under the naive assumption of conditional independence between features.
P(x∣ωj)=P(x1∣ωj)⋅P(x2∣ωj)⋅…⋅P(xn∣ωj)=∏P(xi∣ωj).
□ Dynamics, stability, statistics on lattices & networks: Lyapunov stability analysis for physical/biological problems.
>>
http://scitation.aip.org/content/aip/journal/jmp/55/7/10.1063/1.4881526 …
hydrodynamic description of transport in low spatial dimension, spectral decomposition of stochastic dynamics on directed networks, etc.
□ Quantum Deep Learning: train a deep Boltzmann machine and allow richer classes of models
>>
http://arxiv.org/pdf/1412.3489.pdf …
□ diXa: a Data Infrastructure for Chemical Safety Assessment:
>>
http://bioinformatics.oxfordjournals.org/content/early/2014/12/11/bioinformatics.btu827.full.pdf …










