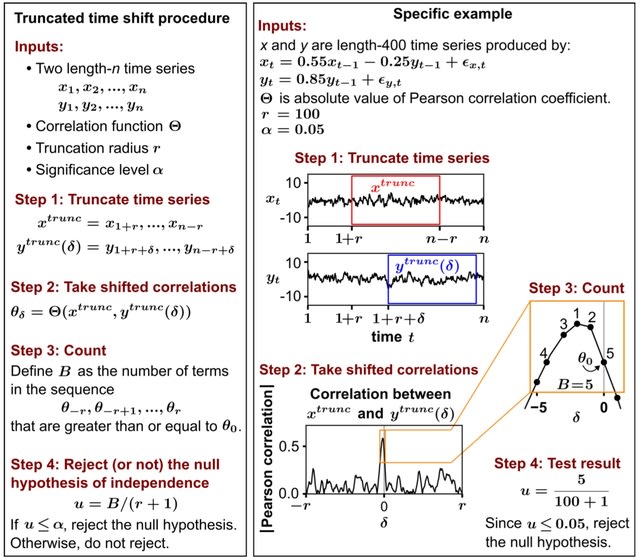
□ Dynamic inference of cell developmental complex energy landscape from time series single-cell transcriptomic data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009821
GraphFP, a nonlinear Fokker-Planck equation (FPE) on graph based model and dynamic inference framework, with the aim of reconstructing the cell state-transition complex potential energy landscape from time series single-cell transcriptomic data.
The discrete Wasserstein distance is introduced to transform the probability simplex into a Riemannian manifold, called discrete Wasserstein manifold. The FPE is proven to be the gradient flow of the free energy on the discrete Wasserstein manifold.
GraphFP learns the complex geometry of data, as well as provides a novel way to quantify cell-cell interactions during cell development. It models the cell developmental process as stochastic dynamics of the cell state/type frequencies on probability simplex in continuous time.

□ End-to-end Learning Of Evolutionary Models To Find Coding Regions In Genome Alignments
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac028/6513381
ClaMSA classifies multiple sequence alignments using a phylogenetic model. It builds on TensorFlow and a custom layer for Continuous-Time Markov Chains (CTMC) and trains a set of rate matrices for a classification task.
This model is the standard general-time reversible (GTR) CTMC that allows to compute gradients of the tree- likelihood under the almost universally used continuous-time Markov chain model.

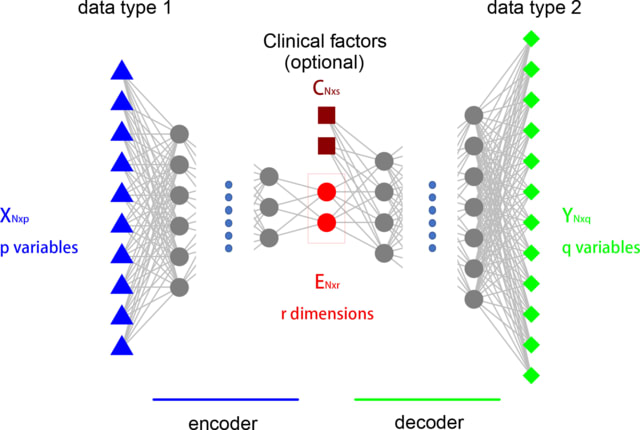
□ DIDL: A deep learning approach to predict inter-omics interactions in multi-layer networks
>>
DIDL is a novel autoencoder architecture that is capable of learning a joint representation of both first-order and second-order proximities. DIDL offers several advantages like automatic feature extraction from raw data, end-to-end training, and robustness to network sparsity.
DIDL is a combination of multilayer perceptron (MLP) and tensor factorization. The predictor and encoder parameters can be jointly optimized. DIDL encoder cluster omics elements through latent feature extraction.

□ LongPhase: an ultra-fast chromosome-scale phasing algorithm for small and large variants
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac058/6519151
LongPhase, an ultra-fast algorithm which can simultaneously phase single nucleotide polymorphisms (SNPs) and SVs of a human genome in ~10-20 minutes, 10x faster than the state-of-the-art WhatsHap and Margin.
LongPhase combined with Nanopore is a cost-effective approach for providing chromosome-scale phasing without the need for additional trios, chromosome-conformation, and single-cell strand-seq data.

□ Syllable-PBWT for space-efficient haplotype long-match query
>> https://www.biorxiv.org/content/10.1101/2022.01.31.478234v1.full.pdf
Syllable-PBWT, a space- efficient variation of the positional Burrows-Wheeler transform which divides every haplotype into syllables, builds the PBWT positional prefix arrays on the compressed syllabic panel, and leverages the polynomial rolling hash function.
The Syllable-Query algorithm finds long matches between a query haplotype and the panel. Syllable-Query is significantly faster than the full memory algorithm. After reading in the query haplotype in O(N ) time, these sequences require O(nβ log M ) time to compute.

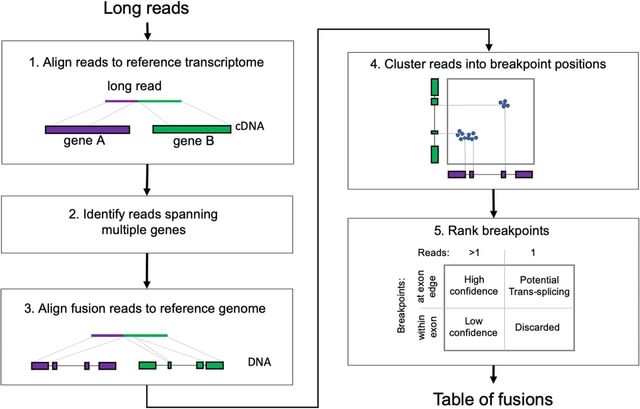
□ VeChat: Correcting errors in long reads using variation graphs
>> https://www.biorxiv.org/content/10.1101/2022.01.30.478352v1.full.pdf
Unlike single consensus sequences, which current approaches are generally centering on, variation graphs are able to represent the genetic diversity across multiple, evolutionarily or environmentally coherent genomes.
VeChat distinguishes errors from haplotype-specific true variants based on variation graphs, which reflect a popular type of data structure for pangenome reference systems.

□ ETNA: Joint embedding of biological networks for cross-species functional alignment
>> https://www.biorxiv.org/content/10.1101/2022.01.17.476697v1.full.pdf
ETNA (Embeddings to Network Alignment) generates individual network embeddings based on network topological structures and then uses a Natural Language Processing-inspired cross-training approach to align the two embeddings using sequence orthologs.
ETNA uses an autoencoder framework to generate lower-dimensional latent embeddings that preserve both local / global network topology while capturing the non-linear relationships. ETNA can be used to transfer genetic interactions across species and identify phenotypic alignments.

□ maxATAC: genome-scale transcription-factor binding prediction from ATAC-seq with deep neural networks
>> https://www.biorxiv.org/content/10.1101/2022.01.28.478235v1.full.pdf
The maxATAC models were specifically designed to improve prediction of TFBS from rare cell types and in vivo settings, where limited sample material or cell sorting strategies would preclude experimental TFBS measurement.
maxATAC predictions for all three TFs outperformed TF motif scanning in ATAC-seq peaks. maxATAC is capable of high resolution TFBS prediction using information-sharing between proximal sequence and accessibility signals.

□ lv89: C implementation of the Landau-Vishkin algorithm
>> https://github.com/lh3/lv89
This repo implements the Landau-Vishkin algorithm to compute the edit distance between two strings. This is a fast method for highly similar strings.
The actual implementation follows a simplified the wavefront alignment (WFA) formulation rather than the original formulation. It also learns a performance trick from WFA.

□ Causal-net category
>> https://arxiv.org/pdf/2201.08963v1.pdf
A causal-net is a finite acyclic directed graph. A category, denoted as Cau, whose objects are causal-nets and morphisms are functors of path categories of causal-nets.
It is called causal-net category and in fact the Kleisli category of the "free category on a causal-net" monad. Cau characterizes interesting causal-net relations, such as coarse-graining, immersion-minor, topological minor, etc., and prove several useful decomposition theorems.

□ Stone Duality for Topological Convexity Spaces
>> https://arxiv.org/pdf/2201.09819v1.pdf
A convexity space is a set X with a chosen family of subsets (called convex subsets) that is closed under arbitrary intersections and directed unions. There is a lot of interest in spaces that have both a convexity space and a topological space structure.
the category of topological convexity spaces and extend the Stone duality between coframes and topological spaces to an adjunction between topological convexity spaces and sup-lattices.
An alternative approach to modelling the category of T0 topological spaces is via strictly zero dimensional biframes. For topological convexity spaces, this construction does not generate any new spaces to improve the properties of the category of spaces.

□ AGAMEMNON: an Accurate metaGenomics And MEtatranscriptoMics quaNtificatiON analysis suite
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02610-4
AGAMEMNON, a time and space-efficient in silico framework for the analysis of metagenomic/metatranscriptomic samples providing highly accurate microbial abundance estimates at genus, species, and strain resolution.
AGAMEMNON uses an EM algorithm to probabilistically resolve the origin of reads. AGAMEMNON takes into account the sparsity of single-cell approaches also in the differential abundance analyses, it offers methods shown to be robust in such settings such as edgeR-LRT and edgeR-QLF.

□ Cross-Dependent Graph Neural Networks for Molecular Property Prediction
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac039/6517516
The multi-view modeling with graph neural network (MVGNN) to form a novel paralleled framework which considers both atoms and bonds equally important when learning molecular representations.
CD-MVGNN, a cross-dependent message passing scheme to enhance information communication of different views. It theoretically justifies the expressiveness of the proposed model in terms of distinguishing non-isomorphism graphs.

□ Discovering adaptation-capable biological network structures using control-theoretic approaches
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009769
Since adaptation is a stable (convergent) response, according to the Hartman–Grobman theorem, the conditions obtained for adaptation using linear time-invariant (LTI) systems theory serve as sufficient conditions for the same even in non-linear systems.
The entire algorithm remains agnostic to the particularities of the reaction kinetics.
The network structures for adaptation ipso facto reduce peak time because of the infinite precision (zero-gain) requirement. The control-theoretic approach addresses the question of non-zero sensitivity along with the infinite precision requirement for perfect adaptation.

□ DeepGOZero: Improving protein function prediction from sequence and zero-shot learning based on ontology axioms
>> https://www.biorxiv.org/content/10.1101/2022.01.14.476325v1.full.pdf
DeepGOZero combines a model-theoretic approach for learning ontology embeddings and protein function prediction. DeepGOZero can exploit formal axioms in the GO to make zero-shot predictions.
DeepGOZero uses a model-theoretic approach for embedding ontologies into a distributed space, ELEmbeddings. ELEmbeddings uses normalized GO axioms as constraints and projects each GO class into an n-ball and each relation as a transformation within n-dimensional space.
DeepGOZero computes the binary crossentropy loss between the predictions and the labels, and optimize them together with four normal form losses for ontology axioms from ELEmbeddings.

□ EagleImp: Fast and Accurate Genome-wide Phasing and Imputation in a Single Tool
>> https://www.biorxiv.org/content/10.1101/2022.01.11.475810v1.full.pdf
EagleImp is 2 to 10 times faster (depending on the single or multiprocessor configuration selected) than Eagle2/Position-based Burrows-Wheeler Transform (PBWT), with the same or better phasing and imputation quality.
EagleImp uses multiple threads for genotype imputation. A conversion of genotypes and haplotypes into a compact representation with integer registers and made extensive use of Boolean and bit masking operations as well as processor directives for bit operations.

□ Lerna: transformer architectures for configuring error correction tools for short- and long-read genome sequencing
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04547-0
Lerna, the automated configuration of k-mer-based EC tools. Lerna first creates a language model (LM) of the uncorrected genomic reads, and then, based on this LM, calculates a metric called the perplexity metric to evaluate the corrected reads for different parameter choices.
Lerna leverages the perplexity metric for automated tuning of k-mer sizes without needing a reference genome. The perplexity computation in Lerna, in contrast, is linear in the length of the input (number of reads × read length). Lerna is 80x to 275x faster than Bowtie2.
Lerna relies on a Simulated Annealing (SA)-based searching. In Lerna, the Transformer LM uses the perplexity metric, which is derived to be the exponential of the cross-entropy loss. Lerna maximizes the similarity between the ground truth and the predictions.

□ GraphChainer: Co-linear Chaining for Accurate Alignment of Long Reads to Variation Graphs
>> https://www.biorxiv.org/content/10.1101/2022.01.07.475257v1.full.pdf
GraphChainer solves co-linear channing on a DAG, when allowing one-node suffix- prefix overlaps between anchor paths. This solution is an extension of the O(k(|V | + |E|) log |V | + kN log N ) time solution. GraphChainer significantly improves the alignments of GraphAligner.
GraphChainer divides the running time of the algorithm into O(k3|V | + k|E|) for pre-processing the graph, and O(kN log kN). That is, for constant width graphs, this solution takes linear time to preprocess the graph plus O(N log N ) time to solve co-linear chaining.

□ MOJITOO: a fast and universal method for integration of multimodal single cell data
>> https://www.biorxiv.org/content/10.1101/2022.01.19.476907v1.full.pdf
MOJITOO uses canonical correlation analysis for a fast detection of a shared representation of cells from multimodal scdata. Moreover, estimated canonical components can be used for interpretation, i.e. association of modality specific molecular features with the latent space.
MOJITOO does not require the definition of parameters such as the rank of the matrix. Furthermore, it provides an approach to estimate the size of the latent space after a single execution of CCA.

□ GAGAM: a genomic annotation-based enrichment of scATAC-seq data for Gene Activity Matrix
>> https://www.biorxiv.org/content/10.1101/2022.01.24.477458v1.full.pdf
Using genes as features solves the problem of the feature dataset dependency allowing for the link of gene accessibility and expression. The latter is crucial for gene regulation understanding and fundamental for the increasing impact of multi-omics data.
a Genomic Annotated GAM (GAGAM), which leverages accessibility data and information from genomic annotations of regulatory regions to weigh the gene activity with the annotated functional significance of accessible regulatory elements linked to the genes.

□ scAR: Probabilistic modeling of ambient noise in single-cell omics data
>> https://www.biorxiv.org/content/10.1101/2022.01.14.476312v1.full.pdf
Single cell Ambient Remover (scAR) which uses probabilistic deep learning to deconvolute the observed signals into native and ambient composition. scAR provides an efficient and universal solution to count denoising for multiple types of single-cell omics data.
This hypothesis suggests that ambient RNAs may not be completely random but deterministic signals to a certain extent.
scAR outputs a probability matrix representing the probability whether raw observed counts contain native signals. scAR simultaneously infers noise ratio (ε) and native expression frequencies (β) using the VAE framework.

□ sc-REnF: An entropy guided robust feature selection for single-cell RNA-seq data
>> https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab517/6509050
sc-REnF [robust entropy based feature (gene) selection method], aiming to leverage the advantages of R′enyi and Tsallis entropies in gene selection for single cell clustering.
sc-REnF raises an objective function that will minimize conditional entropy between the selected features and maximize the conditional entropy between the class label and feature.
While applying sc-REnF multiple times with varying number features, the resulting ARI scores employ a minimum deviation for Renyi and Tsallis entropy.

□ scMVP: A deep generative model for multi-view profiling of single-cell RNA-seq and ATAC-seq data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02595-6
scMVP (the single-cell Multi-View Profiler), a multi-modal deep generative model, which is designed for handling sequencing data that simultaneously measure gene expression / chromatin accessibility in the same cell, incl. SNARE-seq, sci-CAR, Paired-seq, SHARE-seq, and 10X Multiome.
scMVP takes raw count of scRNA-seq and term frequency–inverse document frequency (TF-IDF) transformed scATAC-seq as input.
scMVP automatically learns the common latent representation for scRNA-seq and scATAC-seq data through a clustering consistency-constrained multi-view VAE, and imputes each single layer data from the common latent embedding of the multi-omic data.
scMVP uses a cycle-GAN like auxiliary network. scMVP introduces the multi-heads self-attention module to capture the local long-distance correlation from sparse and high-dimension scATAC profile of joint dataset, and the mask attention to focus on the local semantic region.

□ GPSA: Alignment of spatial genomics and histology data using deep Gaussian processes
>> https://www.biorxiv.org/content/10.1101/2022.01.10.475692v1.full.pdf
Gaussian process spatial alignment (GPSA), a probabilistic model that aligns a set of spatially-resolved genomics and histology slices onto a known or unknown common coordinate system into which the samples are aligned both spatially and in terms of the phenotypic readouts.
GPSA uses two stacked Gaussian processes to align spatial slices across technologies and samples in a two-dimensional, three-dimensional, or potentially spatiotemporal coordinate system. GPSA allows for imputation of missing data and creation of dense spatial readouts.

□ SENIES: DNA Shape Enhanced Two-layer Deep Learning Predictor for the Identification of Enhancers and Their Strength
>> https://ieeexplore.ieee.org/document/9678035/
SENIES is a deep learning based two-layer predictor for enhancing the identification of enhancers and their strength by utilizing DNA shape information beyond two common sequence-derived features, namely kmer and one-hot.
Since there are 7 nucleotide / 6 base pair-step shape parameters used, the length of the concatenated shape feature vector can be formulated as 7×(N−4) + 6×(N−3). Given N=200 in this case, an input DNA sequence can be finally encoded with a DNA shape vector of 2554 dimensions.

□ LuxRep: a technical replicate-aware method for bisulfite sequencing data analysis
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04546-1
LuxRep, a probabilistic method that implements a general linear model and simultaneously accounts for technical replicates (libraries from the same biological sample) from different bisulfite-converted DNA libraries.
LuxRep retains the general linear model with matrix normal distribution used by LuxGLM to handle covariates wherein matrix normal distribution is a generalisation of multivariate normal distribution to matrix-valued random variables.

□ OptiDiff: structural variation detection from single optical mapping reads
>> https://www.biorxiv.org/content/10.1101/2022.01.08.475501v1.full.pdf
OptiDiff uses a single molecule segment-matching approach to the reference map to detect and classify SV sites at coverages as low as 20x. OptiDiff uses a reference molecule set to obtain background mapping levels in all genomic regions on the reference.
OptiDiff calculates the ratio between this background mapping rate and the SV candidate molecules’ mapping rate to detect SV sites. Based on this segment-match information, OptiDiff then applies a simple rule tree to classify the type of structural variation.

□ Illumina
>> https://www.illumina.com/science/genomics-research/articles/infinity-high-performance-long-read-assay.html
The Infinity technology platform combines highly accurate Illumina SBS chemistry, the latest advancements in our data analysis portfolio and a novel proprietary assay to generate long contiguous data to address the most challenging regions of the genome.
Infinity also enables 10x greater throughput with 90% less DNA input than legacy long reads. We anticipate an early access launch for Infinity technology in the second half of the year.

□ Identifying and correcting repeat-calling errors in nanopore sequencing of telomeres
>> https://www.biorxiv.org/content/10.1101/2022.01.11.475254v1.full.pdf
Telomeres are represented by (TTAGGG)n and (CCCTAA)n repeats in many organisms were frequently miscalled (~40-50% of reads) as (TTAAAA)n, or as (CTTCTT)n and (CCCTGG)n repeats respectively in a strand-specific manner during nanopore sequencing.
This miscalling is likely caused by the high similarity of current profiles between telomeric repeats and these repeat artefacts, leading to mis-assignment of electrical current profiles during basecalling.
An overall strategy to re-basecall telomeric reads using a tuned nanopore basecaller. And selective application of the tuned models to telomeric reads led to improved recovery and analysis of telomeric regions, with little detected negative impact on basecalling of other genomic regions.

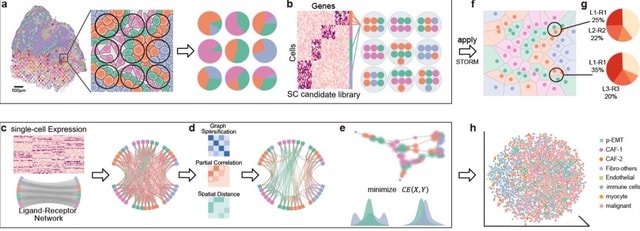
□ CCPLS reveals cell-type-specific spatial dependence of transcriptomes in single cells
>> https://www.biorxiv.org/content/10.1101/2022.01.12.476034v1.full.pdf
CCPLS (Cell-Cell communications analysis by Partial Least Square regression modeling), which is a statistical framework for identifying cell-cell communications as the effects of multiple neighboring cell types on cell-to-cell expression variability of HVGs.
CCPLS performs PLS regression modeling and reports coefficients as the quantitative index of the cell-cell communications. CCPLS realizes a multiple regression approach for estimating the MIMO (multiple-input and multiple-output) system.

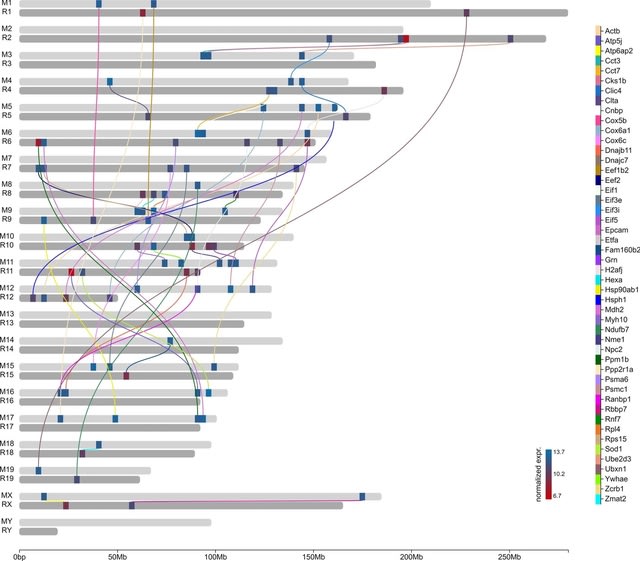
□ The effects of sequencing depth on the assembly of coding and noncoding transcripts in the human genome
>> https://www.biorxiv.org/content/10.1101/2022.01.30.478357v1.full.pdf
the effect of the sequencing depth varied based on cell or tissue type, the type of read considered and the nature and expression levels of the transcripts.
The detection of coding transcripts saturated rapidly for both short-read and long-reads. There was no sign of saturation for noncoding transcripts at any sequencing depth. Increasing long-read sequencing depth specifically benefited transcripts containing transposable elements.

□ HiC-LDNet: A general and robust deep learning framework for accurate chromatin loop detection in genome-wide contact maps
>> https://www.biorxiv.org/content/10.1101/2022.01.30.478367v1.full.pdf
HiC-LDNet can give relatively more accurate predictions in multiple tissue types and contact technologies. HiC-LDNet recovers a higher number of loop calls in multiple experimental platforms, and achieves higher confidence scores in multiple cell types.
HiC-LDNet shows strong robustness when scanning through the extremely sparse scHi-C data, and can recover the majority of the labeled loops. Considering the time complexity, HiC-LDNet could finish its prediction at an average 25s/Mbp across the entire genome at 10kb resolution.

□ NetMix2: Unifying network propagation and altered subnetworks
>> https://www.biorxiv.org/content/10.1101/2022.01.31.478575v1.full.pdf
NetMix2 is an algorithm for identifying altered subnetworks from a wide range of subnetwork families, including the propagation family which approximates the subnetworks ranked highly by network propagation.