









Ein Zeichen sind wir,deutungslos Schmerzlos sind wir und haben fast Die Sprache in der Fremde verloren.
ヘルダーリン『ムネーモシュネー』(第2稿/断章)
個々の事象に与えられる『意味』は、トランザクションの規模によっては変容しない。特定の言語が構造的引力に依って価値を変えるのなら、それは初めから解釈を違えていたか、見え難くなっているかだけのことである。言語間に働く力学構造は、スケールと時系列を横断的にアーキテクトするバベルである。
因果の擬態は相互作用それ自体であるが、音程に重力を感じるなどのシナスタジア(共感覚)が、外界との間に自己相似的な意味を為すか否かは、感覚受容者の認知発火パターンに定量的な相関を認める事とは非同値である。「偶然の符合」と「意味論的な構造」の識別は遠大で、記号する行為は誘導灯である。

『ひとつの記号でわれわれはある、意味もさだかでなく、痛みも知らずにわれわれはあり、異國にあって
ほとんど言葉を失ってしまっている。』
「私」は何者かに成り替わる私であるのではない。私は行為者であるところの「私」であり、「射」それ自体である。四肢が崩れ落ちようとも、軀が朽ちようとも、そのベクトルを失ってはならない。重みさえ置き去りにして周る世界が、留まるものを記憶しようはずがないのだ。

□ Large-Scale Genomic Reorganizations of Topological Domains (TADs) at the HoxD Locus:
>> http://biorxiv.org/content/biorxiv/early/2017/03/12/116152.full.pdf
the re-composition of TADs architectures after severe genomic re-arrangements depends on a boundary-selection mechanism that uses CTCF-mediated gating of long-range contacts in combination with genomic distance and, to a certain extent, sequence specificity.
□ A critical assessment of topologically associating domain (TAD) prediction tools:
>> https://academic.oup.com/nar/article/3059658/A-critical-assessment-of-topologically-associating
Common TAD structure prediction errors included calling smaller TADs by joining consecutive TAD boundaries that may belong to larger meta-TADs or by calling large TADs by joining two consecutive TADs by missing the dividing boundary.

□ HiPiler: Visual Exploration of Large Genome Interaction Matrices with Interactive Small Multiples:
>> http://biorxiv.org/content/biorxiv/early/2017/04/03/123588.full.pdf
integrate the piling metaphor and some of the exploration features from MultiPiles but heavily extend upon them by introducing linear ordering, multi-dimensional arrangements, clustering, filtering, and grouping approaches for exploring many snippets. more seamlessly transition from context-driven (interaction matrix) to knowledge-driven (data attributes) pattern exploration.
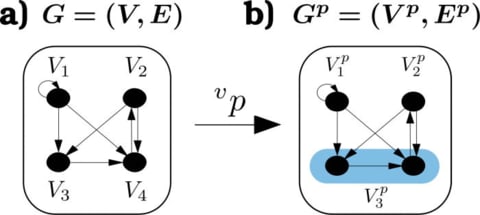
□ Deep graphs: A general framework to represent and analyze heterogeneous complex systems across scales:
>> http://aip.scitation.org/doi/full/10.1063/1.4952963
The framework is based on the ontological assumption that every system can be described in terms of its constituent objects. the mathematical concept of partition lattices is transferred to the network theory in order to demonstrate how partitioning the node & edge set of a graph into supernodes and superedges allows us to aggregate, compute & allocate information on and between arbitrary groups of nodes.
□ ATLAS: a comprehensive infrastructure for assembly and genomic binning of metagenomic and metatranscriptomic data:
>> https://peerj.com/preprints/2843.pdf
ATLAS (Automatic Tool for Local Assembly Structures) provides robust taxonomy based on majority voting of protein-coding open reading frames (ORFs) rolled-up at the contig level using modified lowest common ancestor (LCA) analysis. A DNA-DNA database search module using the Lambda search tool will be added to a future version ATLAS. future versions of ATLAS will include a bin quality control and annotation integrated into our MMVM taxonomic assignment package.

(The model parameters capture meaningful relationships relative to each tensor dimension.)
□ PREDICTD: PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition:
>> http://biorxiv.org/content/biorxiv/early/2017/04/04/123927.full.pdf
the model leverages a machine learning method, tensor decomposition, that holds particular promise in genomics for analyzing increasingly high dimensional data sets. to stabilize the variance across the genome and to more tractable for Gaussian error model, applied an inverse hyperbolic sine transform. imputed data can provide insight into cell type-specific patterns of chromatin state and act as a powerful hypothesis generator.
□ RCAS: an RNA centric annotation system for transcriptome-wide regions of interest:
>> https://academic.oup.com/nar/article/3038237/RCAS-an-RNA-centric-annotation-system-for
The outputs of RCAS can be combined for meta-analysis to compare the characteristics of genomic regions of different biological contexts. The published genomic regions are from the studies using PAR-CLIP, RNA-Seq/deepCAGE and m1A-seq, respectively.
□ Quantum machine learning over infinite dimensions:
>> https://arxiv.org/pdf/1603.06222.pdf
A CV system is characterized by having an infinite-dimensional Hilbert space described by measuring variables w/ a continuous eigenspectra. the vector distance algorithm requires use of a swap test, via the application of the exponential swap gate between two auxiliary states which can be coherent states or squeezed states and the oracle mode and the reference mode.

□ A quantum dynamic belief decision making model:
>> https://arxiv.org/pdf/1703.02386.pdf
The interference effect is measured by the uncertain state in actions and an entanglement degree defined by Deng entropy. the entanglement degree is calculated by an entropy function rather being set as a free parameter.

□ MetaNet: a novel meta learning method that acquires a meta-level knowledge across tasks & shifts its inductive bias.
>> https://arxiv.org/pdf/1703.00837.pdf
MetaNet consists of two main learning components, a base learner and a meta learner, and is equipped with an external memory. A decreasing performance indicates that the meta weights Z and G of the neural nets m and mLSTM are prone to catastrophic forgetting.

□ BRANE Clust: Cluster-Assisted Gene Regulatory Network Inference Refinement:
>> http://biorxiv.org/content/early/2017/03/07/114769
BRANE Clust is a GRN post-processing tool for classical network thresholding refinement. The comparative pertinence of clustering is discussed computationally (SIMoNe, WGCNA, X-means) and biologically (RegulonDB).

□ SciGraph: The new Linked Open Data (LOD) platform Supporting open science and the wider understanding of research:
>> http://www.springernature.com/gp/researchers/scigraph
by the end of 2017 Springer Nature SciGraph will grow to more than one billion triples.
□ Deep and Hierarchical Implicit Models:
>> https://arxiv.org/pdf/1702.08896.pdf
they encompass simulators in the scientific communities, generative adversarial networks, and deep generative models such as sigmoid belief nets and deep latent Gaussian models. These modeling developments could not really be done without inference, and develop a variational inference algorithm that underpins them all.
深層生成モデルや離散データのベイズ生成的対立ネットワークに適応可能な変分推論アルゴリズム.
□ Thar she blows! Ultra long read method for nanopore sequencing
>> http://lab.loman.net/2017/03/09/ultrareads-for-nanopore/
Ultra long reads (up to 882 kb and indeed higher) can be achieved on the Oxford Nanopore MinION using traditional DNA extraction techniques and minor changes to the library preparation protocol, without the need for size selection. a modified Sambrook phenol-chloroform extraction/purification, DNA QC, minimal pipetting steps, high-input to rapid kit and MinKNOW 1.4.

□ Dynamical networks: finding, measuring, and tracking neural population activity using network theory:
>> http://biorxiv.org/content/biorxiv/early/2017/03/10/115485.full.pdf
Typical choices include cosine similarity or a rectified correlation coefficient, these linear measures are familiar, & not data-intensive. But with sufficient data we could also use non-linear measurements of interaction including forms of mutual information & transfer entropy.
□ Modeling Signaling-Dependent Pluripotent Cell States With 2 Boolean Logic Can Predict Cell Fate Transitions:
>> http://biorxiv.org/content/biorxiv/early/2017/03/10/115683.full.pdf
the empirical validation of the SCC approach, where the heterogeneous population of stem cells is assumed to correspond to SCC in the state transition graph of the asynchronously updated Boolean model, has not been performed as it requires live-cell tracking for multi-genes and multi-cells beyond our static cell profiling data collection.
□ Integrating molecular QTL data into genome-wide genetic association: Probabilistic colocalization analysis:
>> http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1006646
further hypothesize that all approaches, including empirical methodologies, make implicit assumptions on the enrichment parameters, which can be understood by the hypothetical example of two perfectly linked SNPs.

□ The authors from deepTools did it again! This time HiCExplorer and HiCBrowser.
>> http://biorxiv.org/content/early/2017/03/08/115063
□ ONT Updates: GridION X5, PromethION, 1D^2, Scrappie, FPGAs and More:
>> http://omicsomics.blogspot.jp/2017/03/ont-updates-gridion-x5-promethion-1d2.html
GridION X5 is a compact benchtop system designed to run and analyse up to five MinION Flow Cells. $125K, $299 per flow cell ($30/Gb at 10G). FPGA Basecalling Accelerator enable to achieve 60% utilization of a 10 teraflop rated device & 7.5Mbases/s calling w/ the same power draw.
□ A cubic-time algorithm for computing the trinet distance between level-1 networks:
>> https://arxiv.org/pdf/1703.05097.pdf
a comparison between the trinet metric and the so-called Robinson-Foulds phylogenetic network metric restricted to level-1 networks. the Robinson-Foulds distance between two trees can be computed in linear time using Day’s algorithm without the need to list all clusters.
□ Evidence of a forward energy cascade & Kolmogorov self-similarity in submesoscale ocean surface drifter observations
>> http://aip.scitation.org/doi/abs/10.1063/1.4974331
The scale-independent energy dissipation rate, or downscale spectral flux, estimated from Kolmogorov’s 4/5th law in this regime closely matches nearby microscale dissipation measurements in the near-surface.
□ Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation
>> http://genome.cshlp.org/content/early/2017/03/15/gr.215087.116.full.pdf
Canu provides graph-based assembly outputs in graphical fragment assembly (GFA) format.
□ HINGE: Long-read assembly achieves optimal repeat resolution:
>> http://genome.cshlp.org/content/early/2017/03/20/gr.216465.116.full.pdf
HINGE allows us to identify 40 datasets where unresolvable repeats prevent the reliable construction of a unique finished assembly. HINGE outputs a visually interpretable assembly graph that encodes all possible finished assemblies consistent with the reads, while other approaches either fragment the assembly or resolve the ambiguity arbitrarily.
□ Distill: a new open science journal and ecosystem supporting human understanding of machine learning.
>> http://distill.pub/about/
>> https://research.googleblog.com/2017/03/distill-supporting-clarity-in-machine.html
Google, OpenAI, DeepMind, and YC have launched their own open science journal, featuring interactive articles.
□ PREDICTING COEXISTENCE IN SPECIES WITH CONTINUOUS ONTOGENETIC NICHE SHIFTS AND COMPETITIVE ASYMMETRY:
>> http://biorxiv.org/content/biorxiv/early/2017/03/22/119446.full.pdf
集団がこれらの選択圧迫にどのように反応するかは、根底にある遺伝的メカニズムの分散および共分散構造を理解することを伴う。 ほとんどの場合、これらの経路を決定する形質の基礎となる遺伝的構造が定量的であり、多数の遺伝子座によって決定されることは圧倒的に高い可能性がある。しかし、単純な個体群統計モデルが異なる遺伝子型間の種内および種間相互作用を記述している集団遺伝学とは異なり、種の生態学的状況における量的特徴の選択(体の大きさ、競合能力、またはニッチシフトなど)について、そのような共生相互作用の結果についての予測をする能力はほとんどない。
□ Disco can do hybrid assembly w PacBio + illumina reads, #JGI2017
>> http://disco.omicsbio.org
□ STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq:
>> http://biorxiv.org/content/biorxiv/early/2017/03/24/120295.full.pdf
defining the truth set as fusions agreed upon anywhere between 2 and 5 methods, ignoring ambiguous or treating ambiguous predictions as FP, and/or considering paralogs as proxies to TP predictions had no effect on STAR-Fusion's top ranking among all methods.
□ PHATE: A Dimensionality Reduction Method for Visualizing Trajectory Structures in High-Dimensional Biological Data:
>> http://biorxiv.org/content/biorxiv/early/2017/03/24/120378.full.pdf
PHATE is a true dimensionality reduction method that preserves heat-diffusion potential distances, such that trajectory structure is naturally and accurately emphasized. PHATE complements methods that extract pseudo-time orderings from data, incl Wanderlust & Wishbone as they can be run on PHATE dimensions.
□ Discovering feature relevancy and dependency by kernel-guided probabilistic model-building evolution:
>> https://biodatamining.biomedcentral.com/articles/10.1186/s13040-017-0131-y
Transforming the input space onto a higher dimensional space using a nonlinear mapping, may resolve nonlinearities w/ linear discriminants. weigthed Kernel Iterative Estimation of Dependencies and Relevancy Algorithm (Kiedra) uses a hybridised version of BMDA & wKiera. The rationale behind the introduction of this criterion is that those nodes with lowest entropy are richer in information content, and thus good candidates to become independent parents of the dependency subnetworks. in this sense this criterion was originally proposed in the MIMIC algorithm.
R𝖬𝖲𝖥 = {rk : rk =arg i min H(Xi ∈ Vk)}
□ Perspective: Energy Landscapes for Machine Learning:
>> https://arxiv.org/abs/1703.07915
Construction of a kinetic transition network also provides a convenient means to visualise a high-dimensional surface. Disconnectivity graphs represent the landscape in terms of local minima and connected transition states, reflecting the barriers and topology through basin connectivity. Energy landscape methods may provide a novel way of addressing one of the most intriguing questions, why does machine learning work so well?
□ BioKanga
>> https://github.com/csiro-crop-informatics/biokanga
BioKanga can process billions of reads against targeted genomes containing 100 million contigs and totalling up to 100Gbp of sequence.
□ Dynamical Scaling and Phase Coexistence in Topologically-Constrained DNA Melting:
>> http://biorxiv.org/content/biorxiv/early/2017/03/29/121673.full.pdf
combining large-scale Brownian Dynamics simulations with an analytically solvable phenomenological Landau mean field theory. a phenomenological Landau mean field theory which couples a critical “denaturation” field (φ) with a non-critical “supercoiling” one (σ).

□ a deep learning paradigm w/ a strong foundation for future improvements in structure-based bioactivity prediction.
>> https://arxiv.org/pdf/1703.10603.pdf
the Atomic Convolutional Neural Network (ACNN) for the purpose of fitting potential energy surfaces, with several key differences which have wide applicability in theoretical chemistry for accelerating ab-initio molecular dynamics simulations of many-body systems.

□ Predicting Causal Relationships from Biological Data: Applying Automated Casual Discovery on Mass Cytometry Data
>> http://biorxiv.org/content/biorxiv/early/2017/03/30/122572.full.pdf
Causal discovery makes assumptions on the nature of causality that connect the observable data properties to the underlying causal structure. automatic causal discovery algorithm typically tries to identify the causal graph that is associated through the Causal Markov assumption.

□ Halvade-RNA: Parallel variant calling from transcriptomic data using MapReduce:
>> https://www.ncbi.nlm.nih.gov/pubmed/28358893?dopt=Abstract
Halvade-RNA configured to run 10 parallel instances of STAR (2 threads) and 20 parallel instances (single threaded) of Picard and GATK, average runtime decreases to ∼3 hours, corresponding to a parallel speedup of 9.18 over sequential execution of the pipeline.