“The OA”NetflixでSeason1を視聴。産業としての映像創作にとって、Genreというものの商業的側面が如何に作家性を犠牲にしてきたのか思いを馳せるチャレンジングな作品。まさに神秘という他ない作品。現代舞踊とイニシエーションを結びつけ、ラストのちゃぶ台返しからのカタルシスは幼少時の頃の創作への高揚感さえ思い出させる。
(165 million year old fossil octopus from France.))
“Life is but a dream full of starts of folly, and of fancied, and true miseries. Death awakes us from this painful dream, and gives us, either a better existence or no existence at all.”
Paradoxは超えられるべき認知上の壁であり、しばしば既知の理論の劇的な転回を伴う。
□ Thresholding normally distributed data creates complex networks:
a simple generative model for continuous valued relational data and study the network ensemble obtained by thresholding it. Despite the simplicity of the model—all the underlying relational data is normally distributed. some, but not all, of the properties associated with complex networks can be seen after thresholding, even though the underlying data is not “complex”.
heavy-tailed degree distributions, large numbers of triangles, and short path lengths, while we do not observe non-vanishing clustering or community structure.
□ ChromA: Characterizing the epigenetic landscape of cellular populations from bulk and single-cell ATAC-seq information:
ChromA, a Bayesian statistical approach that models ATAC-seq information to infer chromatin accessibility landscape and annotate open (accessible) and closed (inaccessible) chromatin regions. ChromA harnesses recent developments in hidden semi-Markov models to create a scalable statistical inference method that can be applied to genome wide experiments.
Nebula utilizes the changes in the count of k-mers to predict the genotype of common structural variations. Nebula relies on CPLEX to generate and solve the linear program. The main phase of the Nebula and the crux of the method is the genotyping stage. Provided the set of k-mers associated with the input SVs, Nebula will count these k-mers in the WGS for genotyping. A final rounding converts the real genotype values to 3 states of 0/0, 0/1 or 1/1.
□ GPseudoClust: deconvolution of shared pseudo-trajectories at single-cell resolution:
GPseudoClust, is a novel approach that both clusters genes for pseudotemporally ordered data and quantifies the uncertainty in cluster allocations arising from the uncertainty in the pseudotime ordering. GPseudoClust combines a recent method for pseudo-time inference with nonparametric Bayesian clustering methods, efficient MCMC sampling, and novel subsampling strategies. For branching data, GPseudoClust identifies differences in dynamic patterns for different branches.
□ cGAUGE: Constraint-based analysis for causal discovery in population-based biobanks:
cGAUGE, a new pipeline for causal Graphical Analysis Using GEnetics that utilizes large changes in the significance of local conditional independencies between the genetic instruments and the phenotypes. search for new graphical patterns to reveal novel information about the underlying causal diagram that is not covered by extant methods, including new direct links, colliders, and evidence for confounding.
□ EpiAlign: an alignment-based bioinformatic tool for comparing chromatin state sequences:
EpiAlign is a dynamic programming al- gorithm that novelly incorporates varying lengths and frequencies of chromatin states. EpiAlign is able to extract recurrent chromatin state patterns along a single epigenome, and many of these patterns carry cell-type-specific characteristics.
□ Genetic and environmental perturbations lead to regulatory decoherence:
The environmental exposures (infection and disease) lead to a systematic loss of correlation, which they define as 'decoherence'. ‘Correlation by Individual Level Product’ (CILP), for each individual the product between two traits is estimated and modeled as a continuous outcome variable.
□ A topological data analysis based classification method for multiple measurement:
a multiple measurement case, take one data point in a tree data set to be a branch of the tree with different features extracted from the Quantitative Structural Modelling (QSM) model. For 6 point process, the alternate SVM classifier appears to maintain accuracy with two of the tree species, while confusing the other four species in a consistent basis, leading to the nearly constant 33% cross-validation accuracy.
This surprising phenomenon possibly reflects a large variation in the data which does not lead to data organization which is accurately partitioned by a hyperplane. Similarly, a potential explanation for the TDA classifier’s high accuracy with more point process classes is that differences in datapoint location in multi-dimensional space could require tools to tease out clusters based on similar geometry.
□ Sequential compression across latent space dimensions enhances gene expression signatures:
Ensembles of features from different unsupervised algorithms (PCA, ICA, non-negative matrix factorization(NMF), denoising autoencoder, and variational autoencoder (VAE) models) discover biological signatures in large transcriptomic datasets. To enhance biological signature discovery, rather than compressing input data into a single pre-selected dimensionality, it is best to perform compression on input data over many latent dimensionalities.
□ Feature Selection and Dimension Reduction for Single Cell RNA-Seq based on a Multinomial Model:
Simple multinomial methods incl. generalized principal component analysis (GLM-PCA) for non-normal distributions, and feature selection using deviance. These methods outperform current practice in a downstream clustering assessment using ground-truth datasets. Dimension reduction via GLM-PCA, or its fast approximation using residuals from a multinomial model, leads to better clustering performance than PCA on z-scores of log-CPM.
The GLM-PCA method is most concordant to the data generating mechanism since all aspects of the pipeline are integrated into a coherent model rather than being dealt with through sequential normalizations and transformations.
□ aphid: an R package for analysis with profile hidden Markov models:
aphid can be used to derive, import and export HMMs and profile HMMs in the R environment. Computationally-intensive dynamic programming recursions such as the Viterbi, forward and backward algorithms are implemented in C ++ and parallelized for increased speed and efficiency.
□ MetaMLP: A fast word embedding based classifier to profile target gene databases in metagenomic samples:
MetaMLP enables partial matching by using a reduced alphabet to build sequence embeddings from full and partial kmers. MetaMLP is able to identify a slightly larger number of reads compared to Diamond as well as to perform accurate predictions with 0.99 precision and 0.99 recall. MetaMLP can process 100M reads in around 10 minutes in a laptop computer which is 50x faster than Diamond.
□ DEPECHE: Determination of essential phenotypic elements of clusters in high-dimensional entities:
DEPECHE, a rapid, parameter free, sparse k-means-based algorithm for clustering of multi- and megavariate single-cell data. In a number of computational benchmarks aimed at evaluating the capacity to form biologically relevant clusters, incl scRNA-seq data w/ manually curated gold standard solutions, DEPECHE clusters as well or better than the currently available best performing clustering algorithms.
□ scSplit: Genotype-free demultiplexing of pooled single-cell RNA-seq:
scSplit uses a hidden state model approach to demultiplex individual samples from mixed scRNA-seq data with high accuracy. This approach does not require genotype information from the indi- vidual samples prior to demultiplexing which also makes it suitable for applications where genotypes are unavailable or difficult to obtain.
□ MultiCapsNet: a interpretable deep learning classifier integrate data from multiple sources:
The single cell RNA sequence data are decoupled into multiple parts according to the structure of MultiCapsNet model that has been integrated with prior knowledge, with each part represents genes influenced by a transcription factor or involved in a protein-protein interaction network and then could be viewed as a data source.
□ Automated exploration of Gene Ontology term and pathway networks with ClueGO-REST:
the GP regression model for analyzing function-valued QTL data, the Bayesian forward selection approach to select the most important markers and the maximum a posterior (MAP) estimate for the hyper-parameters.
The method utilizes approximate Bayesian model posteriors and a stepwise variable selection procedure to efficiently search the model space and find the best subset of molecular markers to be included in the model. This method is non-parametric, includes a minimal number of tuning parameters, and can be applied efficiently to high resolution dynamic data with hundreds of time points.
Dynamic programming matrix for aligning the two linear sequence-to-sequence algorithms to graphs: the Shift-And algorithm for exact matching and Myers’ bitvector algorithm for semi-global alignment. For a graph with |V | nodes and |E| edges and a sequence of length m, the bitvector-based graph alignment algorithm reaches a worst case runtime of O(|V | + [m/w] |E| log w) for acyclic graphs and O(|V | + m|E| log w) for w arbitrary cyclic graphs.
□ Simplifying Genomics Pipelines at Scale with Databricks Delta:
The UAP4G architecture offers flexibility, allowing customers to plug in their own pipelines and develop their own tertiary analytics at petabyte-scale.
□ A Bayesian framework for inferring the influence of sequence context on single base modifications:
a statistical model that allows for the detection and evaluation of the effects of different sequence contexts on mutation rates from deep population sequencing data. the novel Bayesian method based on sparse model selection methods, with the leading assumption that the number of actual sequence contexts that directly influence mutation rates is minuscule compared to the number of possible sequence contexts.
□ ExpansionHunter: A sequence-graph based tool to analyze variation in short tandem repeat regions:
ExpansionHunter uses a novel computational method for genotyping repeats using sequence graphs. This method addresses the long-standing need to accurately genotype medically important loci containing repeats adjacent to other variants or imperfect DNA repeats. ExpansionHunter translates each regular expression into a sequence graph. Informally, a sequence graph consists of nodes that correspond to sequences and directed edges that define how these sequences can be connected together to assemble different alleles.
□ Infinite-dimensional Polish groups and Property (T):
The proof is model-theoretic and does not rely on results of classification of unitary representations. Its main ingredient is the construction, for any א0-categorical metric structure, of a free group on a system of elementary substructures with suitable independence conditions. The theory of the resulting structure is א0-categorical: there is only one separable, infinite-dimensional, complex Hilbert space.
The same applies to the infinite symmetric group, S∞, and to the infinite-dimensional general linear group over the finite field with q elements, GL(∞,Fq).
□ Toward perfect reads: short reads correction via mapping on compacted de Bruijn graphs:
Bcool constructs a compacted de Bruijn graph from the reads. This graph is filtered on the basis of k-mer abundance then of unitig abundance, thereby removing most sequencing errors. The cleaned graph is then used as a reference on which the reads are mapped to correct them. this approach yields more accurate reads than k-mer-spectrum correctors while being scalable to human-size genomic datasets and beyond.
□ Lightweight merging of compressed indices based on BWT variants:
a flexible and lightweight technique for merging compressed indices based on variants of Burrows-Wheeler transform (BWT), thus addressing the need for algorithms that compute compressed indices over large collections using a limited amount of working memory. Starting with a known lightweight algorithm for merging BWTs, they show how to modify it in order to merge, or compute from scratch, also the Longest Common Prefix (LCP) array. and currently working on an improved de Bruijn graph merging algorithm that also supports the construction of succinct Variable Order de Bruijn graph representations.
□ A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes:
a detailed evaluation of reads from Nanopore native RNA sequencing as well as complementary direct cDNA sequencing, from the perspective of transcript identification and quantification. They undertook extensive native RNA sequencing of polyA+ RNA from two human cell lines, and thereby analysed ~5.2 million aligned native RNA reads which consisted of a total of ~4.6 billion bases.
□ Taiyaki: Training models for basecalling Oxford Nanopore reads
Taiyaki is research software for training models for basecalling Oxford Nanopore reads. Oxford Nanopore's devices measure the flow of ions through a nanopore, and detect changes in that flow as molecules pass through the pore. These signals can be highly complex and exhibit long-range dependencies, much like spoken or written language. Taiyaki can be used to train neural networks to understand the complex signal from a nanopore device, using techniques inspired by state-of-the-art language processing.
Taiyaki is used to train the models used to basecall DNA and RNA found in Oxford Nanopore's Guppy basecaller. This includes the flip-flop models, which are trained using a technique inspired by Connectionist Temporal Classification.
□ Deep Learning on Chaos Game Representation for Proteins:
comparing the performance of SVMs, RFs, and DNNs, trained on Frequency matrix Chaos Game Representation (FCGR) encoded protein sequences. While the original chaos game representation (CGR) has been used mainly for genome sequence encoding and classification, they modified it to work also for protein sequences, resulting in n-flakes representation, an image with several icosagons.
□ MALVA: genotyping by Mapping-free ALlele detection of known VAriants:
MALVA computes the set of signatures of length ks of all the alternate alleles of all the variants in VCF and stores them in the set ALTSIG. In the same step, the signatures of the reference alleles are computed and stored in a second set named REFSIG. if a ks-mer of a signature of an alternate allele appears somewhere in the reference genome, MALVA extracts the context of length kc (with kc > ks) covering the reference genome region and collects such kc-mers in a third set (REPCTX).
a probabilistic Count Matrix Factorization (pCMF) approach for single-cell expression data analysis, that relies on a sparse Gamma-Poisson factor model.
Thiis method is competed against other standard representation methods like t-SNE, and we illustrate its performance for the representation of single-cell expression (scRNA-seq) data.
□ Featherweight long read alignment using partitioned reference indexes
Missing mappings refer to primary mappings that were not observed in empirical alignments, but were observed in alignments with reference parameters.
incorporated multi-index merging into the Minimap2 aligner and demonstrate that long read alignment to the human genome can be performed on a system with 2 GB RAM with negligible impact on accuracy.
Minimap2 stands out as the current aligner of choice for long reads, among other long read aligners such as BLASR, GraphMap, Kart, NGMLR and LAMSA; not only is it 30 times faster than existing long read aligners, but its accuracy is on par or superior to other algorithms.
The hash table based approach in Minimap2 has been shown to be effective for long reads. In contrast, FM-index based short read aligners such as BWA and Bowtie have been shown to fail with ultra long reads.
□ SMAUG: Analyzing single-molecule tracks with nonparametric Bayesian statistics
SMAUG provides a mathematically rigorous approach to measuring the real-time dynamics of molecular interactions in living cells.
a new approach to analyzing single-molecule trajectories: the Single-Molecule Analysis by Unsupervised Gibbs sampling (SMAUG) algorithm, which uses nonparametric Bayesian statistics to uncover the whole range of information contained within a single-particle trajectory (SPT) dataset.
the violation of the second law via a device called a Maxwell's demon," Lesovik said. "The most recent paper approaches the same problem from a third angle: artificially created a state that evolves in a direction opposite to that of the thermodynamic arrow of time."
□ gnomAD-SV: An open resource of structural variation for medical and population genetics
gnomAD-SV, a reference atlas of SVs from deep WGS in ~15,000 samples aggregated as part of gnomAD. A rich landscape of 498,257 unique SVs, including 5,729 multi-breakpoint complex SVs across 13 mutational subclasses, and examples of localized chromosome shattering, like chromothripsis, in the general population.
And calculate that 87% of SVs across all populations were in Hardy-Weinberg Equilibrium, although this is an imperfect metric given the potential confounding assumptions and population genetic forces that may not hold true for all SV sites.
□ Sci-Hi-C: a single-cell Hi-C method for mapping 3D genome organization in large number of single cells
by combining HiCRep with multidimensional scaling (MDS), we was able to implement an analytical tool to embed sci-Hi-C data into a low- dimensional space, which successfully separate cell subtypes from a cell population based on cell-to-cell variations in cell-cycle phase.
□ Matrix factorization for multivariate time series analysis
the factorization provides a decomposition of each series in a dictionary which member that can be interpreted as latent factors used for example in state-space models.
Estimation by empirical risk minimization. By multiplying both sides by the pseudo-inverse Λ+ = Λ∗(ΛΛ∗)−1, we obtain the “simplified model”
The rows of ε are independent and have the same T-dimensional are derived. sub-Gaussian distribution, with second moment matrix Σε.
□ HiLDA: a statistical approach to investigate differences in mutational signatures
a hierarchical latent Dirichlet allocation model (HiLDA) for characterizing somatic mutation data that allows the user to simultaneously extract mutational signatures and infer mutational exposures between two different groups of mutational catalogs.
HiLDA provides posterior distributions for each parameter, thereby allowing construction of 95% credible intervals for parameters.
□ Primo: integration of multiple GWAS and omics QTL summary statistics for elucidation of molecular mechanisms of trait-associated SNPs and detection of pleiotropy in complex traits
Primo identifies SNPs in various association patterns to complex and omics traits and performs conditional association analysis in a region to account for linkage disequilibrium.
the conditional association analysis of Primo examines the conditional associations of a SNP with multiple traits adjusting for other lead SNPs in a gene region. It moves beyond joint association towards causation and colocalization, and provides a thorough inspection of the effects of multiple SNPs within a region to reduce spurious associations due to LD.
In Deep Medicine, leading physician Eric Topol reveals how artificial intelligence can help. AI has the potential to transform everything doctors do, from notetaking and medical scans to diagnosis and treatment, greatly cutting down the cost of medicine and reducing human mortality.
“the only way it can be made secure is to be decentralized.”
"Machine medicine need not be our future.”
Eric Topol著 『DEEP MEDICINE』が届いたので、入院のお供に。ディープラーニングを応用した最新医学の潮流を展望する内容。電子版でも購入済みだけれど、一つの記念碑として図書を購入。共有もし易いしね😎✨
“The triad of deep phrnotyping - knowing more about the person’s layers of medical data than was ever previously attainable or even conceived - deep learning and deep empathy can be major remedy to the economic crisis.”
“DeepSequence and DeepVariant are AI tools to understand the functional effect mutations or make accurate calls of genomic variants, respetively, used to predict transcriotion factors.” Topol. “DEEP MEDICINE” p.212
“BenevolentAI”, one of the largest private AI companies in Europe, has built natural language processing that shifts through the biomedical literature and chemical databases.”
“The idea that intellectuall tasks can be categorized as automatable grunt work will probably be insulting to many chemists, and will certanity feel like a threat.”
“But the use of artificial intelligence will actually free up time in which they can think more about higher-level questions of which molecules should be made and why, rather than focusing on the details of how to make molecules.”
Terra is part of The Data Biosphere: a growing community bringing data and life science together. Terra is product of The Broad Institute of MIT & Harvard Data Sciences Platform in collaboration with Verily Life Sciences.
Terra includes a well-stocked repository of hundreds of best practice, ready-for-use bioinformatics workflows, and an easy interface with linked third-party applications (e.g. Dockstore, FireCloud, Jupyter Notebooks).
□ Mapping DNA replication with nanopore sequencing
The ionic conductivity through the nanopore is particularly sensitive to the nucleobases located in its narrowest region. Thus, changes in the ionic current levels during translocation reveal changes in the DNA sequence.
Several consecutive nucleobases in the narrowest region can influence the ionic current. Translating a sequence of current values into a DNA sequence is therefore a non-trivial task. Several machine learning approaches using either Hidden Markov Models (HMM) or Recurrent Neural Networks (RNN) have been developed by ONT or by academic groups to reach this goal.
□ Accurate detection of complex structural variations using single-molecule sequencing
introduce open-source methods for long-read alignment NGMLR and structural variant identification Sniffles that provide unprecedented sensitivity and precision for variant detection, even in repeat-rich regions and for complex nested events that can have substantial effects.
NGMLR and Sniffles can automatically filter false events and operate on low-coverage data, thereby reducing the high costs that have hindered the application of long reads in clinical and research settings.
□ New Characterizations for the Multi-output Correlation-Immune Boolean Functions
In stream ciphers, the correlation-immune functions serve as an important met- ric for measuring a cryptosystem resistance against correlation attacks. In block ciphers, there is a equivalence between multipermutations and correlation-immune functions.
three new methods are proposed to characterize the multioutput correlation-immune Boolean functions. Compared with the Walsh spectral characterization method, the first method can significantly reduce the computational complexity. The last two characterization methods are in terms of the Fourier spectral characterizations over the complex field.
The new method of describing multi-output correlation-immune Boolean functions in terms of discrete Fourier transform over the complex field.
□ A Bayesian model for single cell transcript expression analysis on MERFISH data
Multiplexed error-robust fluorescence in-situ hybridization (MERFISH) is a recent technology to obtain spatially resolved gene or transcript expression profiles in single cells for hundreds to thousands of genes in parallel.
The presented model is implemented on top of Rust-Bio and available open-source as MERFISHtools. this enables MERFISH to scale towards the whole genome while being able to control the uncertainty in obtained results.
□ Efficient parameterization of large-scale dynamic models based on relative measurements
a novel hierarchical approach combining the efficient analytic evaluation of optimal scaling, offset, and error model parameters with the scalable evaluation of objective function gradients using adjoint sensitivity analysis.
□ Higher order analysis of gene correlations by tensor decomposition
A new fast algorithm to formulate a genes × genes × genes decorrelated rank-1 sub-tensors (complexes) which can be associated with functionally independent pathways.
□ Is evolution predictable? Quantitative genetics under complex genotype-phenotype maps
To overcome the limitations of LASSO, elastic net, and existing boosting methods, they develop a novel method, termed Integrative Boosting (I-Boost), which combines elastic net with boosting. In I-Boost, the prediction rule is constructed iteratively, where at each iteration, the predictive power of each data type is evaluated separately and the most predictive data type is selected to update the prediction rule using elastic net.
□ PrivMCMC: Protecting Genomic Data Privacy with Probabilistic Modeling
This is achieved by combining minimal amounts of perturbation with Bayesian statistics and Markov Chain Monte Carlo techniques.
PrivMCMC takes in genotype and phenotype data, and allows a use to estimate the privacy loss by releasing statistics related to that data. Here, the loss of privacy is measured in terms of posterior probabilities.
□ The Beacon Calculus: A formal method for the flexible and concise modelling of biological systems
the beacon calculus, a process algebra designed to simplify the task of modelling in- teracting biological components. Its breadth is demonstrated by creating models of DNA replication dynamics, the gene expression dynamics in response to DNA methylation damage, and a multisite phosphorylation switch.
EpiSAFARI employs a novel smoothing method for decreasing noise in signal profiles and accounts for technical factors such as sparse signals, mappability, and nucleotide content. In performances e comparisons, EpiSAFARI performs favorably in terms of accuracy.
EpiSAFARI computes the significance of the depletion at the dip compared to the summits. evaluated several background models for assigning the significance. The multi-mappability, nucleotide content, hill score and statistical significance are used to filter the valleys.
□ Deep Learning with Multimodal Representation for Pancancer Prognosis Prediction
an unsupervised encoder to compress these four data modalities into a single feature vector for each patient, handling missing data through a resilient, multimodal dropout method. Encoding methods were tailored to each data type - using deep highway networks to extract features from genomic and clinical data, and convolutional neural networks.
use of multimodal data, novel representation learning techniques, and methods such as multimodal dropout to create models that can generalize well and predict also in the absence of one or more data modalities.
□ Characterizing human genomic coevolution in locus-gene regulatory interactions
characterize coevolution as it appears in locus-gene interactions in the human genome, focusing on expression Quantitative Trait - Locus (eQTL) interactions.
Pairs that appear coevolved are enriched for intra-chromosomal regulatory interactions, thus our results suggest that measures of coevolution can be useful for prediction and detection of new interactions.
Measures of coevolution are genome-wide and could potentially be used to prioritize the detection of distant or inter-chromosomal interactions such as trans-eQTL interactions in the human genome.
If A is a finite CW-complex such that algebraic theories detect mapping spaces out of A, then A has the homology type of a wedge of spheres of the same dimension. Furthermore, if A is simply connected then A has the homotopy type of a wedge of spheres.
the class of detectable spaces is larger if we consider also infinite CW-complexes. Spheres localized at a set of primes are detectable.
□ pysradb: A Python package to query next-generation sequencing metadata and data from NCBI Sequence Read Archive
pysradb that provides a collection of command line methods to query and download metadata and data from SRA utilizing the curated metadata database available through the SRAdb project.
pysradb enables seemless downloads from NCBI’s SRA. It organizes the downloaded data following NCBI’s hiererachy: ‘SRP => SRX => SRR’ of storing data. Each ‘SRP’ (project) has multiple ‘SRX’ (experiments) and each ‘SRX’ in turn has multiple ‘SRR’ (runs).
□ The tricategory of formal composites and its strictification
The results of this thesis allow one to replace calculations in tricategories with equivalent calculations in Gray categories (aka semistrict tricategories).
consider for an arbitrary tricategory T atricategory T^ of "formal composites", which distinguishes (formal) composites of 1- or 2-morphisms, even if they evaluate to the same 1-resp. 2-morphism in T.
Then T^ is strictly triequivalent -that means triequivalent via a strict functor of tricategories- to a tricategory Tst. This result, together with the observation that T^ is strictly triequivalent to T, is crucial for reducing calculations in tricategories to calculations in Gray categories.
□ High-dimensional nonparametric density estimation via symmetry and shape constraints
the K-homothetic log-concave maximum likelihood estimator based on n independent observations from such a density has a worst-case risk bound with respect to, e.g., squared Hellinger loss, of O(n−4/5), independent of p.
the estimator is adaptive in the sense that if the data generating density admits a special form, then a nearly parametric rate may be attained.
This estimation algorithms are fast even when n and p are on the order of hundreds of thousands, and we illustrate the strong finite-sample performance of our methods on simulated data.
□ Network meta-analysis correlates with analysis of merged independent transcriptome expression data
Merging of high-dimensional expression data can, however, implicate batch effects that are sometimes difficult to be removed.
demonstrated in a simulation study and by the analysis of a real-world example that network meta-analysis is a useful tool to make additional inferences from multiple independent studies with high-dimensional molecular expression data.
□ Distributed Detection with Empirically Observed Statistics
A binary distributed detection problem in which the distributions of the sensor observations are unknown and only empirically observed statistics are available to the fusion center.
In most practical distributed detection systems, the local decision rule at each sensor is a deterministic compressor or quantizer. However, under certain conditions, randomized local decision rules can be used to provide privacy or to satisfy power constraints.
maximize the type-II error exponent over the proportions of channels for both source and training sequences and conclude that as the ratio of the lengths of training to test sequences tends to infinity, using only one channel is optimal.
□ The Truth and Nothing but the Truth: Multimodal Analysis for Deception Detection
a data-driven method for automatic deception detection in real-life trial data using visual and verbal cues.
a lexical analysis on the spoken words, emphasizing the use of pauses and utterance breaks, feeding that to a Support Vector Machine to test deceit or truth prediction.
□ A haplotype-aware de novo assembly of related individuals using pedigree graph, apparently more accurate and contiguous than TrioCanu
This method is cost- effective, requiring only 30× short-read coverage and 15× long-read coverage for every individual in a pedigree to generate near chromosomal- scale assemblies for all individuals.
These assemblies for both real and simulated data are more accurate and contiguous when compared to those produced by TrioCanu at 45× child long-read coverage.
□ A complete statistical model for calibration of RNA-seq counts using external spike-ins and maximum likelihood theory
The method is derived by maximum likelihood theory in the context is a complete statistical model for sequencing counts contributed by cellular RNA and spike-ins.
adopted a multinomial mode for spike-in noise, but this model could be extended with a more accurate model. maximum likelihood normalization method can be improved, with respect to clustering and detection of differential gene expression, by applying an RUVr.
□ DDOT: A Swiss Army Knife for Investigating Data-Driven Biological Ontologies
The Data-Driven Ontology Toolkit (DDOT) facilitates the inference, analysis, and visualization of biological hierarchies using a data structure called an ontology.
Using DDOT, they programmatically assemble a compendium of ontologies for 652 diseases by integrating gene-disease mappings with a gene similarity network derived from omics data.
□ TipToft: detecting plasmids contained in uncorrected long read sequencing data
Given some raw uncorrected long reads, such as those from PacBio or Oxford Nanopore, predict which plasmid should be present. Assemblies of long read data can often miss out on plasmids, particularly if they are very small or have a copy number which is too high/low when compared to the chromosome.
TipToft gives you an indication of which plasmids to expect, flagging potential issues with an assembly.
A higher depth of read coverage will increase the power to accurately identify plasmid sequences, the level of which depends on the underlying base error rate.
For sequence data with 90% base accuracy, an approximate depth of read coverage of 5 is required to identify a plasmid replicon sequence with 99.5% confidence.
□ Evolutionary dynamics in structured populations under strong population genetic forces
characterize a regime in which selection and migration interact to create non-monotonic patterns of the population differentiation statistic FST when migration is weaker than selection, but stronger than drift.
demonstrate how these patterns can be leveraged to estimate high migration rates that would lead to panmixia in the long term equilibrium using an approximate Bayesian computation approach.
□ scScope: Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning
scScope, a scalable deep-learning-based approach that can accurately and rapidly identify cell-type composition from millions of noisy single-cell gene-expression profiles.
A major innovation of scScope is the design of a self-correcting layer. This layer exploits a recurrent network structure to iteratively perform imputations on zero-valued entries of input scRNA-seq data.
□ Cell composition analysis of bulk genomics using single-cell data
Cell Population Mapping (CPM), a deconvolution algorithm in which reference scRNA-seq profiles are leveraged to infer the composition of cell types and states from bulk transcriptome data.
Analysis of individual variations reveals that the relationship between cell abundance and clinical symptoms is a cell-state-specific property that varies gradually along the continuum of cell-activation states.
□ Increasing the Transparency of Research Papers with Explorable Multiverse Analyses
extended the Tangle library to support additional widgets, such as the choice lists from the example Dataverse.
In Freqentist, Dataverse and Dance, all possible figures were generated in R and exported as bitmaps with a naming scheme allowing JavaScript to retrieve the figure corresponding to a specific combination of analysis options.
Coordinated views and linking could be used to facilitate navigation in the multiverse, for example by highlighting the currently visible analysis in a multiverse summary.
□ Bifrost: Highly parallel construction and indexing of colored and compacted de Bruijn graphs
a comparative assessment of the different k-mer frequency estimation programs (ntCard, KmerGenie, KmerStream and Khmer (abundance-dist-single.py and unique-kmers.py) to assess their relative merits and demerits.
The results indicate that ntCard is more accurate in estimating F0, f1 and full k-mer abundance histograms compared with other methods. ntCard is the fastest but it has more memory requirements compared to KmerGenie.
□ How sequence alignment scores correspond to probability models
Pair-wise alignment is traditionally based on ad hoc scores for substitutions, insertions, and deletions, but can also be based on probability models (pair hidden Markov models: PHMMs).
The best way to score alignments and assess their significance depends on the aim: judging whether whole sequences are related versus finding related parts. This clarifies the statistical basis of sequence alignment.
□ HyperPhylo: Data Distribution for Phylogenetic Inference with Site Repeats via Judicious Hypergraph Partitioning
Using empirical MSA data, these sites-to-core assignments computed via HyperPhylo are substantially better than those obtained via a previous na ̈ıve approach for phylogenetic data distribution under SRs.
HyperPhylo, a highly efficient and scalable open-source implementation for a specific flavor of judicious hypergraph partitioning where every vertex has the same degree. intend to integrate this into our RAxML tool for maxmimum likelihood based phylogenetic inference.
□ CGAT-core: a python framework for building scalable, reproducible computational biology workflows
CGAT-core seamlessly handles parallelisation across high performance computing clusters, integration of Conda environments, full parameterisation, database integration and logging.
To illustrate CGAT-core workflow framework, present a pipeline for the analysis of RNAseq data using pseudo-alignment.
□ Multiplatform Biomarker Identification using a Data-driven Approach Enables Single-sample Classification
The Data-Driven Reference (DDR) approach, which employs stably expressed housekeeping genes as references to eliminate platform-specific biases and non-biological variabilities.
The categories generated by stably expressed reference genes represent "built-in features that have a consistent interpretation across gene expression platforms and eliminate sample-specific biases.
□ Relative Lempel-Ziv and FUSE: Compressed Filesystem for Managing Large Genome Collections
The system obtains orders-of-magnitude compression by using Relative Lempel-Ziv, which exploits the high similarities between genomes of the same species.
The filesystem transparently stores the files in compressed form, intervening the system calls of the applications without the need to modify them.
This prototype is a proof of concept to show that relative compression specifically designed for genomic data can be applied in a filesystem. It was designed to use RLZ for sequence data, but the capability of separating metadata text from fasta files and compressing it with LZMA.
□ Gene activation precedes DNA demethylation in response to infection in human dendritic cells
LNISKS, a mutation discovery method which is suited for large and repetitive crop genomes. In this experiments, it rapidly and with high confidence, identified mutations from over 700 Gbp of bread wheat genomic sequence data.
□ On triangular inequalities of correlation-based distances for gene expression profiles
Propose an alternative dr to the absolute correlation distance, defined as dr = √(1 − |ρ|), where ρ can be Pearson correlations, Spearman correlations, or Cosine similarity. The clustering method includes hierarchical clustering and PAM (partitioning around medoids).
Further investigate droplet-based single-cell RNA-seq platforms in terms of zero-inflation, use of negative control data, where absolutely no biological heterogeneity is expected. This will answer whether technical shortcomings in scRNA-seq methods produces an excess of zeros compared to expectations.
1. Elemental Trigger
2. Synergistic Perceptions
3. Hidden Refuge
4. Delusion Fields
5. Omnipresent Boundary
6. Undulating Terrain
7. A Point of No Return
a guide to possible interpretations of an ambivalent, a non-euclidean geometry, as yet unmapped, inaccessible through linear perceptions. to illuminate, decode and decipher this landscape of fractured density, ultimately to deconstruct these unfolding dimensions, where dreams are only whispers.
□ Stephen Turner (@strnr):
I spent a few hours with Gödel Escher Bach this weekend. Not really getting into it, but I hate giving up on books I've started. Help me out here. Should I power through? Is GEB really that good?
□ Clive G. Brown (@Clive_G_Brown)
Nobody ever finishes it.
PathRacer is a novel standalone tool that aligns profile HMM directly to the assembly graph (performing the codon translation on fly for amino acid pHMMs) by modern assemblers in standard Graphical Fragment Assembly (GFA) format. PathRacer provides the set of most probable paths traversed by a HMM through the whole assembly graph, regardless whether the sequence of interested is encoded on the single contig or scattered across the set of edges, therefore significantly improving the recovery of sequences of interest even from fragmented metagenome assemblies.
□ The complexity dividend: when sophisticated inference matters:
constructing a hierarchy of strategies that vary in complexity between these limits and find a power law of diminishing returns: increasing complexity gives progressively smaller gains in accuracy. Moreover, the rate at which the gain decrements depends systematically on the statistical uncertainty in the world, such that complex strategies do not provide substantial benefits over simple ones when uncertainty is too high or too low. In between, when the world is neither too predictable nor too unpredictable, there is a complexity dividend.
□ Accurate inference of tree topologies from multiple sequence alignments using deep learning:
a deep convolutional neural network (CNN) to infer quartet topologies from multiple sequence alignments. This CNN can readily be trained to make inferences using both gapped and ungapped data. The confidence scores produced by this CNN can more accurately assess support for the chosen topology than bootstrap and posterior probability scores. This approach is highly accurate and is remarkably robust to bias-inducing regions of parameter space such as the Felsenstein zone and the Farris zone.
□ Omnipresent Maxwell's demons orchestrate information management in living cells:
A core gene set encoding these functions belongs to the minimal genome required to allow the construction of an autonomous cell. These Maxwell's demon (MxD) allow the cell to perform computations in an energy‐efficient way that is vastly better than our contemporary computers.
□ synder: inferring genomic orthologs from synteny maps:
Ortholog inference is a key step in understanding the evolution and function of a gene or other genomic feature. Yet often no similar sequence can be identified, or the true ortholog is hidden among false positives. A solution is to consider the sequence’s genomic context. synder is the generic program for tracing features of interest between genomes based on a synteny map.
□ SViCT: Structural variation and fusion detection using targeted sequencing data from circulating cell free DNA:
SViCT can detect breakpoints and sequences of various structural variations including deletions, insertions, inversions, duplications and translocations. SViCT extracts discordant read pairs, one-end anchors and soft-clipped/split reads, assembles them into contigs, and re-maps contig intervals to a reference genome using an efficient k-mer indexing approach. The intervals are then joined using a combination of graph and greedy algorithms to identify specific structural variant signatures.
□ An MCMC-based method for Bayesian inference of natural selection from time series DNA data across linked loci:
This approach relies on a hidden Markov model incorporating the two-locus Wright-Fisher diffusion with selection, which enables us to explicitly model genetic recombination. The posterior probability distribution for selection coefficients is obtained by using the particle marginal Metropolis-Hastings algorithm, which also allows for co-estimation of the population haplotype frequency trajectories.
□ SCANVIS: a tool for SCoring, ANnotating and VISualing splice junctions:
SCANVIS generates reasonable PSI scores by demonstrating that tissue/cancer types in GTEX and TCGA are well separated and easily predicted from a few thousand SJs. SCANVIS scores and annotates SJs with attention to many details that can assist in the inference of any downstream consequences. It runs quite efficiently.
□ Learning Topology: topological methods for unsupervised learning:
"Topology gives a different way on classical problems. Dimensional Reduction, Clustering, Anomaly Detection, everything is in one pocket."
Data is uniformly distributed on the manifold. Define a Riemannian metric on the manifold to make this assumption true. Topology and category theory provide a different language to frame problems.
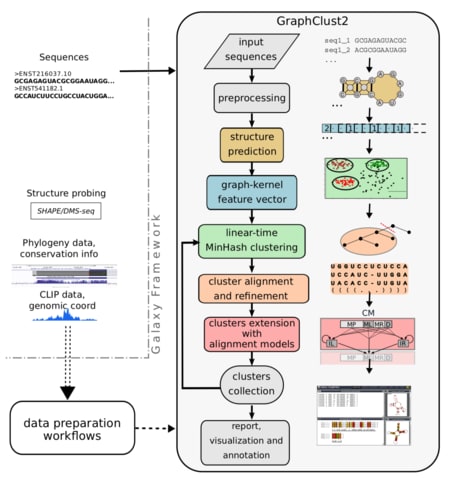
□ GraphClust2: Empowering the annotation and discovery of structured RNAs with scalable and accessible integrative clustering:
The GraphClust methodology uses a graph kernel approach to integrate both sequence and structure information into high-dimensional sparse feature vectors. hese vectors are then rapidly clustered, with a linear-time complexity over the number of sequences, using a locality sensitive hashing technique.
The linear-time alignment-free methodology of GraphClust2 , accompanied by cluster refinement and extension using RNA comparative methods and structure probing data.
□ BRIDGE: Biomolecular Reaction & Interaction Dynamics Global Environment:
the foundations of BRIDGE developed on the Galaxy platform that makes possible fundamental molecular dynamics of proteins through workflows and pipelines via commonly used packages such as NAMD, GROMACS and CHARMM. BRIDGE can be used to set up and simulate biological macromolecules, perform conformational analysis from trajectory data and conduct data analytics of large scale protein motions using statistical rigor.
□ KrakenLinked: Novel Algorithms for the Taxonomic Classification of Metagenomic Linked Reads:
If reads from the same long fragment of DNA are identified via assembly (e.g. Athena) or by barcode deconvolution (e.g. Minerva) the markers from both reads could be used together for taxonomic classification. Read-clouds provide useful additional taxonomic information even without clearly identifying which reads originated from the same fragment. A purpose built Linked-Read taxonomic classifier could search for a parsimonious set of taxonomic assignments for each read cloud.
□ Highly Efficient Hypothesis Testing Methods for Regression-type Tests with Correlated Observations and Heterogeneous Variance Structure:
As fast and numerically stable replacements for the weighted LMER test, the PB-transformed tests are especially suitable for “messy” high-throughput data that include both independent and matched/repeated samples. By using this method, the practitioners no longer have to choose between using partial data or ignoring the correlation in the data.
□ IRIS-EDA: An integrated RNA-Seq interpretation system for gene expression data analysis:
IRIS-EDA, which is a Shiny web server for expression data analysis. Seven discovery-driven methods (correlation analysis, heatmap, clustering, biclustering, Principal Component Analysis, Multidimensional Scaling, and t-SNE are provided for gene expression exploration. IRIS-EDA provides a framework to expedite submission of data and results to NCBI’s Gene Expression Omnibus following the FAIR (Findable, Accessible, Interoperable and Reusable) Data Principles.
□ EpiAlignment: alignment with both DNA sequence and epigenomic data:
EpiAlignment takes DNA sequence and epigenomic profiles derived by ChIP- seq, DNase-seq, or ATAC-seq from two species as input data, and outputs the best semi-global alignments. These alignments are based on EpiAlignment scores, computed by a dynamic programming algorithm that accounts for both sequence alignment and epigenome similarity.
□ Novel-X: Detection and assembly of novel sequence insertions using Linked-Read technology:
Novel-X detects and genotypes novel sequence insertions in 10X sequencing dataset using non-trivial read alignment signatures and barocde information. In the case of SuperNova assembler, user has no control for any assembler parameters & default parameters are maximally tuned for whole human genome diploid assembly. A future direction would be to develop a specific local assembler designed for SV detection in Linked-Read data.
□ ReorientExpress: Transcriptome long-read orientation with Deep Learning:
ReorientExpress is a tool to predict the orientation of cDNA reads from error-prone long-read sequencing technologies. It was developed with the aim to orientate nanopore long-reads from unstranded cDNA libraries without the need of a genome or transcriptome reference, but it is applicable to any set of long-reads. It builds kmer-based models using Deep Neural Networks using as training input a transcriptome annotation or any other fasta/fasq file for which the sequences orientation is known.
□ High-Fidelity Nanopore Sequencing of Ultra-Short DNA Sequences:
a nanopore-based sequencing strategy in which short target sequences are first circularized and then amplified via rolling-circle amplification to produce long stretches of concatemeric repeats. These can be sequenced on the Oxford Nanopore Technology’s (ONT) MinION platform, and the resulting repeat sequences aligned to produce a highly-accurate consensus that reduces the high error-rate present in the individual repeats.
□ Fastq-pair: efficient synchronization of paired-end fastq files:
The algorithm creates a hash of objects that contain the sequence identifier and the position in the file for all the identifiers in the first file. This significantly reduces the memory requirements for fastq-pair compared to trivial solutions that store sequence identifier. fastq-pair provides a rapid and memory efficient solution, written in C for portability, that synchronizes paired-end fastq files for subsequent analysis and places unmatched reads into singleton files.
DNA- and RNA-like systems built from eight nucleotide “letters” (hence the name “hachimoji”) that form four orthogonal pairs. Has normal A, T, G, and C, plus new bases of B, Z, P, and S. 8-letter DNA can also be transcribed using an engineered T7 RNA polymerase.
These synthetic systems meet the structural requirements needed to support Darwinian evolution, including a polyelectrolyte backbone, predictable thermodynamic stability, and stereoregular building blocks that fit a Schrödinger aperiodic crystal.
a Fubini rule for ∞-co/ends of ∞-functors F : Co × C → D. This allows to lay down “integration rules”, similar to those in classical co/end calculus, also in the setting of ∞-categories. the definition of twisted arrow ∞-category of an ∞-category; in this paves the way to the definition of co/end for a ∞-functor F : Co × C → D. in the ∞-category of spaces, the object (C,C′)/Σ exhibits the same universal property of MapC(C,C′).
□ Unbiased estimation of linkage disequilibrium from unphased data:
an approach to compute unbiased estimators for a broad set of two-locus statistics, for either phased or unphased data. This includes commonly used statistics, such as D and D2, the additional statistics in the Hill-Robertson system (D(1 − 2p)(1 − 2q) and p(1 − p)q(1 − q)), and, in general, any statistic that can be expressed as a polynomial in haplotype frequencies (f’s) or in terms of p, q, and D.
□ Latent Space Phenotyping: Automatic Image-Based Phenotyping for Treatment Studies:
Beyond computational requirements, another limitation of the method is a substantial difference in interpretability compared to GWAS using standard image-based phenotyping techniques. the measured phenotype can be directly interpreted as the biomass of the sample and QTL can be found which correlate with the effect of the treatment on biomass. candidate loci obtained through Latent Space Phenotyping must be interpreted differently.
□ Cooler: scalable storage for Hi-C data and other genomically-labeled arrays:
Cooler is a sparse data model and file format for genomically-labelled arrays with minimal redundancy but enough flexibility to support a wide range of data types, data sizes and future metadata requirements. A sparse representation in particular is crucial for developing robust tools and algorithms for use on increasingly high-resolution multi-dimensional genomic data sets that need to operate on subsets of data at a time.
□ A semi-parametric Bayesian approach, iSBA, for differential expression analysis of RNA-seq data:
modeling the RNA-seq count data with a Poisson-Gamma mixture model, and propose a Bayesian mixture modeling procedure with a Dirichlet process as the prior model for the distribution of fold changes between the two treatment means. Markov chain Monte Carlo (MCMC) posterior simulation using Metropolis Hastings algorithm to generate posterior samples for differential expression analysis while controlling false discovery rate.
□ CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing:
CHETAH (CHaracterization of cEll Types Aided by Hierarchical clustering) is an accurate cell type identification algorithm that is rapid and selective, including the possibility of intermediate or unassigned categories. For cells of an unknown type, CHETAH is more selective, yielding a classification that is as fine-grained as is justified by the available data.
□ SCIRA: Leveraging high-powered RNA-Seq datasets to improve inference of regulatory activity in single-cell RNA-Seq data:
SCIRA (Single Cell Inference of Regulatory Activity), which leverages the power of large-scale bulk RNA-Seq datasets to infer high-quality tissue-specific regulatory networks, from which regulatory activity estimates in single cells can be subsequently obtained.
Power-calculation for SCIRA/SEPIRA: the strategy to estimate the sensitivity of SEPIRA/SCIRA to detect highly expressed cell-type specific TFs in a given tissue, as a function of the corresponding cell-type proportion in the tissue.
□ AKRON: Approximate kernel reconstruction for time-varying networks:
AKRON supersedes the Lasso regularization by starting from the Lasso-Kalman inferred network and judiciously searching the space for a sparser solution that is more representative of the ground truth. The results demonstrate that the proposed approach is better at recovering sparse time-varying networks than l1-KF. Not only was the reconstruction error of the proposed approach lower than l1-KF, but it was also better at detecting whether an edge exists in a network.
□ ACValidator: a novel assembly-based approach for in silico validation of circular RNAs:
ACValidator is a bioinformatics approach to perform in silico validation of selected circular RNA junction(s). ACValidator operates in three phases: extraction of reads from SAM file, generation of a “pseudo-reference” file, and assembly and alignment of extracted reads. ACValidator extracts reads from a user-defined window on either side of the circRNA junction and assembles them to generate contigs. These contigs are aligned against the circRNA sequence to find contigs spanning the back-spliced junction.
□ CausalTAB: the PSI-MITAB 2.8 updated format for signalling data representation and dissemination:
a common standard for the representation and dissemination of signalling information: the PSI Causal Interaction tabular format (CausalTAB) which is an extension of the existing PSI-MI tab-delimited format, now designated PSI-MITAB 2.8. As the causal regulatory mechanism is taken from the PSI-MI ontology, the database name is 'psi-mi'. When the ‘Interaction Types’ column is equal to the psi-mi term: psi-mi:"MI:2286", then the causal regulatory mechanism must be absolutely filled - otherwise it can be empty.
□ The Linked Selection Signature of Rapid Adaptation in Temporal Genomic Data:
develop theory that predicts the magnitude of these temporal autocovariances, showing that it is determined by the level of additive genetic variation, recombination, and linkage disequilibria in a region. Furthermore, by using analytic expressions for the temporal variances and autocovariances in allele frequency, demonstrate one can estimate the additive genetic variation for fitness and the drift-effective population size from temporal genomic data.
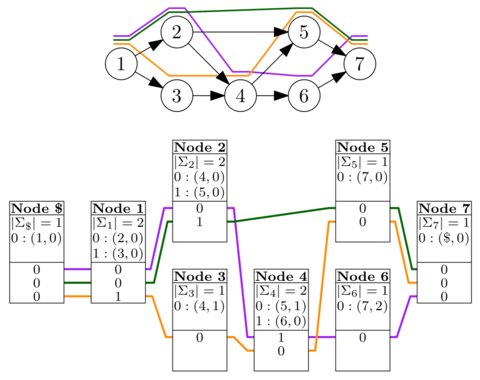
GBWT, a scalable implementation of the graph extension of the positional Burrows–Wheeler transform, to store the haplotypes as paths in the graph. Haplotypes are essentially sequences of nodes in the variation graph, and GBWT is best seen as the multi-string BWT of the node sequences. VG graphs are connected, we can use the long-range information in the GBWT for mapping long reads and paired-end reads.
□ FQSqueezer: k-mer-based compression of sequencing data:
QSqueezer, a novel compression algorithm for sequencing data able to process single- and paired-end reads of variable lengths. It is based on the ideas from the famous prediction by partial matching and dynamic Markov coder algorithms.
□ Massive computational acceleration by using neural networks to emulate mechanism-based biological models:
With this framework, we can predict not only the 1-D distribution in space (partial differential equation models) and probability density function (stochastic differential equation models) of variables of interest with high accuracy, but also novel system dynamics not present. the NN is more likely to fail when the system output is highly sensitive to local parameter variations, i.e., where the output landscape is rugged in the parametric space.
□ SCINA: Semi-Supervised Analysis of Single Cells in silico:
SCINA, a semi-supervised model, for analyses of scRNA-Seq and flow cytometry/CyTOF data, and other data of similar format, by automatically exploiting previously established gene signatures using an expectation–maximization (EM) algorithm. The average correct assignment probability of the total of 2000 cells was 98.44% for SCINA, 52.50% for KC, 52.72% for SINCERA and 18.46% for PhenoGraph, which indicated that SCINA achieved a much more stable performance than unsupervised clustering.
□ A component overlapping attribute clustering (COAC) algorithm for single-cell RNA sequencing data analysis and potential pathobiological implications:
a network-based approach, termed Component Overlapping Attribute Clustering (COAC), to infer novel gene-gene subnetworks in individual components (subsets of whole components) representing multiple cell types and phases of scRNA-seq data. COAC can reduce batch effects and identify specific cell types in two large-scale human scRNA-seq datasets.
□ Graph Peak Caller: Calling ChIP-seq peaks on graph-based reference genomes:
Graph Peak Caller is based on the same principles used by MACS2, and is able to call peaks with or without a set of control alignments. For the case of a graph that merely reflects a linear reference genome, our peak-caller produces the same results as MACS2.
□ R-DECO: An open-source Matlab based graphical user interface for the detection and correction of R-peaks:
Within R-DECO, the R-peaks are detected by an adaptation of the Pan-Tompkins algorithm. Instead of using all the pre-processing steps of the latter algorithm, the proposed algorithm uses only an envelope-based procedure to flatten the ECG and enhance the QRS-complexes.
□ DeepSV: Accurate calling of genomic deletions from high throughput sequencing data using deep convolutional neural network:
DeepSV is based on a novel method of visualizing sequence reads. The visualization is designed to capture multiple sources of information in the sequence data that are relevant to long deletions.
□ ANEVA-DOT prioritized a gene in a patient that led to a new diagnosis. A pseudoexon insertion caused by an intronic variant that resulted in NMD. continue incorporating it into the Mendelian RNA-seq workflow.
ANEVA estimates of regulatory variation correlate with other estimates of constraint (GTEx v7 data, 10k RNA-seq samples spanning 48 tissues and 620 individuals, total 14k genes used, mostly protein-coding genes) #grd19
The General Meeting brings together technology, software, applications, data resources and public policy. AGBT provides a forum for exchanging information about the latest advances in DNA sequencing technologies, experimental and analytical approaches for genomic studies, and their myriad applications.
Visium Spatial Gene Expression Solution
・Examine histological and transcriptome profiles from the same tissue section with our new and improved solution
・Obtain unbiased and high-throughput gene expression analysis for intact tissue sections in a wide variety of samples types
・Unravel the biological architecture by understanding the interplay of collections of localized cells in normal and diseased tissue
Chromium Connect
・Streamline your single cell library preparation and maximize efficiency with our automated system
・Obtain consistent single cell gene expression results by minimizing variability across users
・Operate with ease using a touchscreen computer, for new and experienced users
□ mason_lab:
Karen Miga from @UCSC showing how to get 11 reference-quality genomes in 9 days on @nanopore PromethION, using @circulomics short read eliminator kit to get N50 at 44kb for reads #AGBT19
@mike_schatz routinely getting >100GB per @nanopore flowcell on the PromethION w/ @CSHL @shgoodwin1 -structural variant detection with crossstitch
Sedlazeck, Rescheneder, et al (2018) @naturemethods . doi:10.1038/s41592-018-0001-7 showing new tandem duplications
Mikhail Kolmogorov showing the new, impressive genome assembler Flye: #AGBT19 , including reconstruction of the telomeres and other regions of tandem repeats. Code here: https://github.com/fenderglass/Flye
□ adampeltan:
Jennifer Ishii from NY Genome Center talking about @NEBiolabs new enzymatic alternative to bisulfite sequencing em-seq #AGBT19
□ Tuuli Lappalainen: @tuuliel
Giving a talk tomorrow afternoon on our new methods (w @Pejminister) for modeling regulatory variation and improving diagnosis with allelic expression.
□ Nick Loman: @pathogenomenick
A new platform ! It’s been a while. Genapsys is ‘iPad sized’, short read electronic sequencer (100bp?) with Illumina-like accuracy for $10k. Launching later this year. #AGBT19
□ BariBallew:
Dovetail Genomics at #AGBT19 has a very cool method of recovering reasonable length reads from FFPE DNA. Turns cross-linked DNA from a hurdle to a critical part of the protocol.
□ A prototype of a blockchain-based clinical trial management system that ensures immutability and traceability of trial data
□ DaleYuzuki:
Hunkapillar #AGBT19 Using Google DeepVariant increased accuracy doing this calling a different way.
□ WillSalerno:
#AGBT19 M. Hunkapiller @PacBio: CCS adds 156,105 variants to GIAB reference set, improving medically relevant regions, phasing.
I say again, clinical completeness means "edge cases": high F-measure isn't good enough. Would love to see more weird AF reference sets.
@PacBio showing CCS "Correctness" vs GIAB better than PB CLR and ONT+-Illumina. SV deletion GIAB recall ~96% vs ~76% with Manta. CCS+GATK good SNVs, less good indels. Deep Variant model trained on CCS: big improvements, especially indels.
□ acarroll_ATG:
Clairvoyante uses an end-for-end ML approach (vs the candidate generation process of DeepVariant). The high class imbalance makes this a challenging ML task, but may help for long, noisy reads. It also uses a smaller network than DV, which means it can run very quickly.
□ Clairvoyante: A multi-task convolutional deep neural network for variant calling in single molecule sequencing
Clairvoyante, a multi-task five-layer convolutional neural network model for predicting variant type (SNP or indel), zygosity, alternative allele and indel length from aligned reads.
Skyhawk mimics how a human visually identifies genomic features comprising a variant and decides whether the evidence supports or contradicts the sequencing read alignments using the network architecture they developed in a previous study named "Clairvoyante". Clairvoyante generates only one tensor for each variant candidate. It could be beneficial for calling variants from noisy reads.
Clairvoyante generates only one tensor for each variant candidate. It could be beneficial for calling variants from noisy reads. In contrast, the more rigorous candidate generation and labeling process used in DeepVariant with a larger deep learning network may be essential for its best-in-class performance for high-quality data (e.g. read error rate.
□ DaleYuzuki:
Rodrigues #AGBT19 NGS-compatible tools for studying dynamics, perhaps 1,000 or 10,000 cells. How to get dynamic information? Not neurosci-specific. A few technologies: pulse-chase; derivative; pseudo-time; recorders.
□ Generating high-quality human reference genomes using PromethION nanopore sequencing:
- Avg. ~7x ultralong (100kb+/22GB) reads from 3 flowcells each, for 11 human genomes
- At peak, running 15 flow cells concurrently (2.4TB in 9 days)
- Weighted allele analysis convincing to focus now on quality not quantity of genomes for pangenome
□ RachelGoldfeder:
Long read sequencing is getting a lot of attention at #AGBT19 this year! Learn more about these technologies at @jacksonlab in CT this Sept - looking forward to hearing the latest awesome science from @mason_lab @mike_schatz @pathogenomenick and others!
□ aphillippy:
I am excited to announce the open release of 50x coverage @nanopore ultra-long data of the CHM13 human cell line, and the first gapless assembly of a human X chromosome! Work in progress with @khmiga to finally finish the human reference. Data:
□ longreadclub:
Adam Phillippy talking about #longreadclub and how you can get ultra long reads - get in touch with us and join the long read club community #AGBT19
□ khmiga:
I am thrilled to share the announcement of our Telomere-to-Telomere (T2T) Consortium and open release of 50x coverage of UL nanopore sequence. Come hear @aphillippy talk on this and first release of the first gapless assembly of the X Chromosome! #AGBT19
The Telomere-to-Telomere (T2T) consortium is an open, community-based effort to generate the first complete assembly of a human genome.
the CHM13hTERT human cell line on the Oxford Nanopore GridION. sequenced approximately 50x coverage using 10X Genomics as well as BioNano DLS and Arima Genomics HiC. PacBio data for this cell line has been previously generated by the Washington University School of Medicine and the University of Washington, and is available from NCBI SRA.
Human genomic DNA was extracted from the cultured cell line. As the DNA is native, modified bases will be preserved. We followed Josh Quick's ultra-long read (UL) protocol for library preparation and sequencing.
"The assembly is 2.94 Gbp in size with 657 contigs and an NG50 of 85.8 Mbp."
Done with @Scalene's ultra-long read protocol.
#LongReadClub @longreadclub
□ taylorlawley:
A complete human genome is coming in the next 2 years @aphillippy @khmiga #AGBT19 Like, REALLY complete. @longreadclub @oxfordnanopore
□ illuminaLive:
KP: Shape + sequence motifs can overlap, extend each other. Shape recognition is common in co-bound regions, as an alternate to protein "tethering". Shape motifs might be thought of as complex sequence motif with dependent sites. #AGBT19
A De Novo Shape Motif Discovery Algorithm Reveals Preferences of Transcription Factors for DNA Shape Beyond Sequence Motifs
□ GS: Demonstrating the small but mighty accurate power of iSeq, improvements on NextSeq550 flow cells and with extended array capabilities, and finally the new offering of SPrime flow cell on NovaSeq:
□ DRAGEN (Dynamic Read Analysis for GENomics) v3.2.8:
DRAGEN includes high-quality SNV calling with Manta, and Repeat Expansion detection with ExpansionHunter v1.0. The sequencing systems generate binary base calls (BCLs), which must be demultiplexed into FASTQs (read + quality score) for secondary analysis.
□ Longshot: accurate variant calling in diploid genomes using single-molecule long read sequencing:
Longshot harnesses long SMS reads to jointly perform SNV detection and haplotyping. For this, it uses the read-based haplotype phasing method HapCUT2. To overcome the high error rate of SMS reads, it utilizes a pair-Hidden Markov Model to average over the uncertainty in the local alignments and estimate accurate base quality values that can be used for calculating genotype likelihoods.
□ minimap2 v2.16 released with faster ultra-long nanopore mapping, option to output unmapped reads in PAF and a few bug fixes. Available at bioconda, PyPI (for mappy) and github:
□ ewanbirney:
Lif scaleable ... means the full genome dimension and spatial dimension open to study for RNA in organs. Only need time dimension. The full 5D space over an organism (3D, time, genome) means —- “just” efficient sampling and the mother of all imputations.
Slide-seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution
instead of directly aggregating peaks based on existing genomic annotations, Destin adopts weighted PCA, with peak-specific weights calculated based on the distances to TSSs as well as the relative frequency of chromatin accessibility peaks based on reference regulomic data. The weights, the hyperparameters, as well as the number of principle components, are cast as tuning parameters and are determined based on the likelihood calculated from a post-clustering multinomial model.
□ Avant-garde: An automated data-driven DIA data curation tool:
Avant-garde is a new tool to refine DIA (Data-Independent Acquisition) and PRM by removing interfered transitions, adjusting integration boundaries and scoring peaks to control the FDR. Unlike other tools where MS runs are scored independently from each other, Avant-garde uses a novel data-driven scoring strategy. DIA signals are refined by learning from the data itself, using all measurements in all samples together to achieve the best optimization.
□ Deep learning in bioinformatics: introduction, application, and perspective in big data era:
they provide eight examples, which cover five research directions, four data types, and a number of deep learning models that people will encounter in Bioinformatics. The five research directions are: sequence analysis, structure prediction and reconstruction, biomolecular property and function prediction, biomedical image processing and diagnosis, biomolecule interaction prediction and systems biology. The four data types are: structured data, 1D sequence data, 2D image or profiling data, graph data. The covered deep learning models are: deep fully connected neural networks, ConvNet, RNN, graph convolutional neural network, ResNet, GAN, VAE.
□ Mash Screen: High-throughput sequence containment estimation for genome discovery:
The MinHash algorithm has proven effective for rapidly estimating the resemblance of two genomes or metagenomes. However, this method cannot reliably estimate the containment of a genome within a metagenome. Mash Screen, an online algorithm capable of measuring the containment of genomes and proteomes within either assembled or unassembled sequencing read sets.
□ fragilityindex: An R Package for Statistical Fragility Estimates in Biomedicine:
The fragility index was introduced as a convenient measure to estimate how fragile statistical results in clinical trials are to small perturbations in event outcome counts. extended the concept of a statistical fragility index to two of the most commonly used methods in clinical research, survival analysis via weighted log-rank tests and logistic regression, and implemented these technique sin this R package.
□ SpaRC: scalable sequence clustering using Apache Spark:
SparkReadClust (SpaRC), that partitions reads based on their molecule of origin to enable downstream assembly optimization. SpaRC produces high clustering performance on transcriptomes and metagenomes from both short and long read sequencing technologies. SpaRC provides a scalable solution for clustering billions of reads from next-generation sequencing experiments, and Apache Spark represents a cost-effective solution with rapid development/deployment cycles for similar large-scale sequence data analysis problems.
□ Megabase Length Hypermutation Accompanies Human Structural Variation at 17p11.2
using long- and short-read sequencing technologies to investigate end products of de novo chromosome 17p11.2 rearrangements and query the molecular mechanisms underlying both recurrent and non-recurrent events. single-stranded DNA is formed during the genesis of the SV and provide compelling support for a microhomology-mediated break-induced replication (MMBIR) mechanism for SV formation.
□ CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps
Cluster analysis begins with the HDBSCAN algorithm to conservatively segment the datasets into regions containing clusters and a matrix or noise region. The stability of each cluster and the probability that an atom belongs to a cluster are quantified. Each clustered region is further analyzed by the DeBaCl algorithm to separate and refine clusters present in the sub-volumes. Finally, the k-nearest neighbor algorithm may be used to re-assign matrix atoms to clusters, based on their probability values.
□ Assemblosis: Common Workflow Language (CWL)-based software pipeline for de novo genome assembly from long- and short-read data:
The workflow first extracts, corrects, trims and decontaminates the long reads. Decontaminated trimmed reads are then used to assemble the genome and raw reads are used to polish it. Next, Illumina reads are cleaned and used to further polish the resultant assembly. Finally, the polished assembly is masked using inferred repeats and haplotypes are eliminated.
Using the CWL-based assembly workflow, all three genome assemblies completed successfully. Metrics from the evaluation methods Quast and Genome Assembly Gold-standard Evaluations (GAGE) were used to compare the CWL-based assemblies to respective, high quality reference genomes.
□ pycoQC, interactive quality control for Oxford Nanopore Sequencing"
Gene Set Information Analysis derives entropy, interaction information and mutual information for gene sets on interaction networks, starting from a simple phenomenological model of a living cell. Formally, the model describes a set of interacting linear harmonic oscillators in thermal equilibrium. Because the energy function is a quadratic form of the degrees of freedom, entropy and all other derived information quantities can be calculated exactly.
□ Ephemeral persistence modules and distance comparison:
A key feature of persistence theory is that the various versions of the space of persistent modules can be endowed with pseudo-distances. a definition of ephemeral multi-persistent modules and prove that the quotient of persistent modules by the ephemeral ones is equivalent to the category of γ- sheaves. In the case of one-dimensional persistence, our definition agrees with the usual one showing that the observable category and the category of γ-sheaves are equivalent, and compare the interleaving and convolution distances.
□ Higher topological complexity of aspherical spaces:
The topological complexity, TC(X), informally, is the minimal number of continuous rules which are needed to construct an algorithm for autonomous motion planning of a system having X as its configuration space. The quantity TC(X) is a numerical homotopy invariant of a path-connected topological space and may be studied with all the tools of algebraic topology. A survey of the concept TC(X) and robot motion planning algorithms in practically interesting configuration spaces can be found.
□ On the Time Complexity of Algorithm Selection Hyper-Heuristics for Multimodal Optimisation:
For a function class called CLIFFd where a new gradient of increasing fitness can be identified after escaping local optima, the hyper-heuristic is extremely efficient while a wide range of established elitist and non-elitist algorithms are not, incl. the Metropolis algorithm. complete the picture with an analysis of another standard benchmark function called JUMPd as an example to highlight problem characteristics where the hyper-heuristic is inefficient.
□ Nanopore sequencing of long ribosomal DNA amplicons enables portable and simple biodiversity assessments with high phylogenetic resolution across broad taxonomic scale:
assemble a pipeline for long rDNA barcode analysis and introduce a new software (MiniBar) to demultiplex dual indexed nanopore reads, and find excellent phylogenetic and taxonomic resolution offered by long rDNA sequences across broad taxonomic scales. Sequencing dual indexed, long rDNA amplicons on the MinION platform is a straightforward, cost effective, portable and universal approach for eukaryote DNA barcoding.
□ Enhancing the prediction of disease-gene associations with multimodal deep learning:
As an integrative model, multimodal deep belief net (DBN) can capture cross-modality features from heterogeneous datasets to model a complex system. a joint DBN is used to learn cross-modality representations from the two sub-models by taking the concatenation of their obtained latent representations as the multimodal input. Finally, disease-gene associations are predicted with the learned cross-modality representations.
□ absSimSeq: Compositional Data Analysis is necessary for simulating and analyzing RNA-Seq data:
a normalization approach that uses negative control features w/ log-ratios, which called compositional normalization, and created an extension of sleuth, sleuth-ALR to use compositional normalization or RUVSeq, both to predict candidate reference genes and to normalize the data. Log-ratio transformations are undefined if either the numerator or denominator is zero. Sleuth-ALR has implemented an imputation procedure for handling zeros that minimizes any distortions on the dependencies within the composition.
□ MIA: Daniel Kunin, Jon Bloom, Aleks Goeva, Cotton Seed. Topology of regularized linear autoencoders
a visual introduction to Morse theory, which relates the topology (shape) of a manifold (space) to the behavior of smooth, real-valued functions on that manifold. the Morse homology of Euclidean space forces geometric relationships between critical points, establishing a theoretical foundation for fast geometric ensembling that in turn suggests new algorithms. For the algebraic topologists: we realized squared distance from a k-plane to a point cloud in R^m is an F2-perfect Morse function on the real Grassmannian. This gave intuition for the LAE thm which in turn proved the abstract thm in coordinates.
□ Loss Landscapes of Regularized Linear Autoencoders:
L2-regularized LAEs learn the principal directions as the left singular vectors of the decoder, providing an extremely simple and scalable algorithm for rank-k SVD. Overall, this work clarifies the relationship between autoencoders and Bayesian models and between regularization and orthogonality. a simple, biologically-plausible, and testable resolution of the "weight symmetry problem" namely a local mechanism by which maximizing information flow & minimizing energy expenditure gives rise to backpropagation as the optimization algorithm underlying efficient neural coding.
"Real-world networks are often said to be ”scale free”. @annadbroido and @aaronclauset test this claim on real-world networks, showing that scale-free structure is far from universal."
Mathematical results based on extreme degree heterogeneity may, in fact, have more narrow applicability than previously believed, given the lack of evidence that empirical moment ratios diverge as quickly as those results typically assume. The variation of evidence across social, biological, and technological domains is consistent with a general conclusion that no single universal mechanism explains the wide diversity of degree structures found in real-world networks.
□ bin3C: exploiting Hi-C sequencing data to accurately resolve metagenome-assembled genomes:
bin3C is an open-source pipeline and makes use of the Infomap clustering algorithm. Infomap is another clustering algorithm, which like Markov clustering is based upon flow. Rather than considering the connectivity of groups of nodes versus the whole, flow models consider the tendency for random walks to persist in some regions of the graph longer than others.
□ ARMOR: an Automated Reproducible MOdular workflow for preprocessing and differential analysis of RNA-seq data:
ARMOR (Automated Reproducible MOdular RNA-seq) is a Snakemake workflow, aimed at performing a typical RNA-seq workflow in a reproducible, automated, and partially contained manner.
Directors: Ethan Coen, Joel Coen
Writers: Joel Coen, Ethan Coen
Stars: Tim Blake Nelson, Willie Watson, Clancy Brown
Six tales of life and violence in the Old West, following a singing gunslinger, a bank robber, a traveling impresario, an elderly prospector, a wagon train, and a perverse pair of bounty hunters.
1. Dance of the Evil Toys
2. Conversation Among the Ruins
3. Snake Hip Waltz
4. Cianna
5. Nilaste
6. Life Filtering from the Water Flowers
7. The Windup
The Secret Between the Shadow and the Soul finds the celebrated ensemble at a new peak, addressing a kaleidoscope of moods with inspiration and group commitment. The quartet that saxophonist Branford Marsalis has led for the past three decades has always been a model of daring, no-apologies artistry, of ever-widening musical horizons and deepening collective identity.
現代ジャズの最高峰と称えられるブランフォード・マルサリス・カルテットの新譜"The Secret Between the Shadow and the Soul"から。寂しげな曲調の中にも、細やかな陰翳を刻む饒舌なサックスが味わい深い。










 (165 million year old fossil octopus from France.))
(165 million year old fossil octopus from France.))