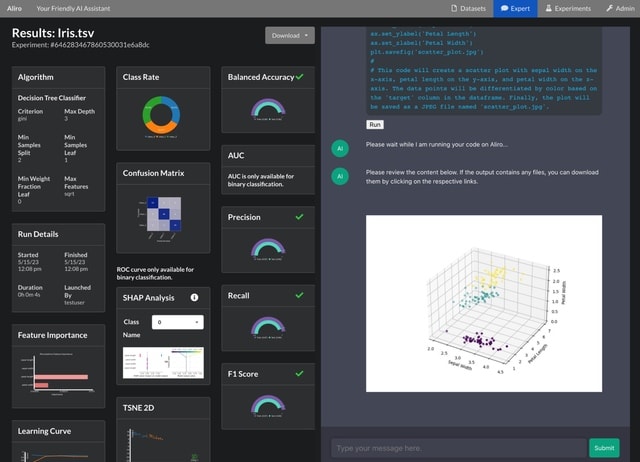
□ scDiff: A General Single-Cell Analysis Framework via Conditional Diffusion Generative Models
>> https://www.biorxiv.org/content/10.1101/2023.10.13.562243v1
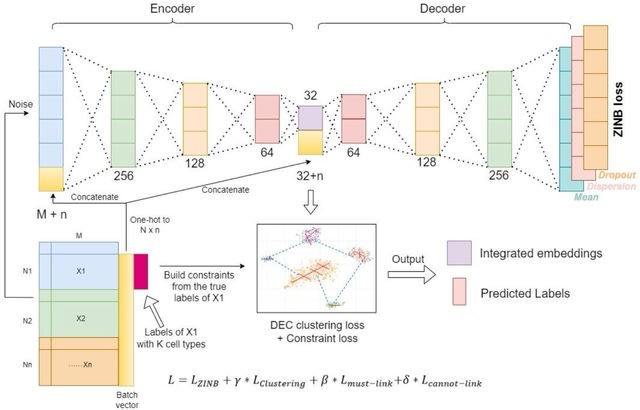
scDiff enables extensive conditioning strategies. Besides LLMs and GNNs, we can enhance scDiff with other guidance methods, like CLIP. scDiff can be promptly extended to multiomics or multi-modality tasks.
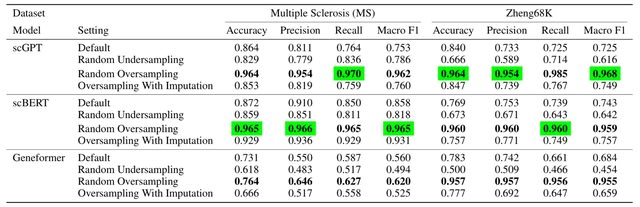
scDiff uses a conditional diffusion generative model to approximate the posterior by a Markov chain. scDiff shows outstanding few-shot and zero-shot results. scDiff outperforms GEARS among all the metrics and datasets except the MSE on Norman.


□ SecDATA: Secure Data Access and de novo Transcript Assembly protocol - To meet the challenge of reliable NGS data analysis
>> https://www.biorxiv.org/content/10.1101/2023.10.26.564229v1
SecDATA, an optimized pipeline for de novo transcript assembly that adopts a Blockchain-based strategy. The major focus here lies towards implementing (a) a pipeline that accesses secured data with the help of DLT and (b) performs de novo transcript sequence reconstruction.
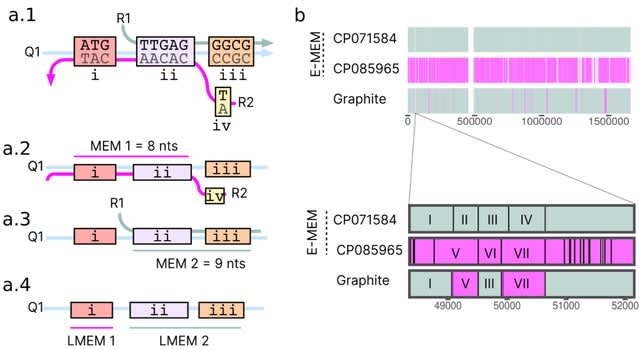
The "Optimized length" represents the minimum number of nodes traversed for building all transcripts i.e. minimum path length for transcript construction. SecDATA uses overlaps in k-mers to determine which k-mer pairs are adjacent in the read sequences.
SecDATA uses Ethereum techniques. SecDATA encompasses blocks or nodes, which are connected through a network. The nodes communicate through a secure channel and use the hash value as a key.

□ DeepGenomeVector: Towards AI-designed genomes using a variational autoencoder
>> https://www.biorxiv.org/content/10.1101/2023.10.22.563484v1
DeepGenomeVector can learn the basic genetic principles underlying genome composition. In-depth functional analysis of a generated genome vector suggests that it is near-complete and encodes largely intact pathways that are interconnected.
DeepGenomeVector involves training a generative variational autoencoder, consisting of three layers, with a latent representation size of 100 neurons. The model was trained to optimize the sum of binary cross-entropy loss and Kullback-Leibler divergence.

□ Deep DNAshape: Predicting DNA shape considering extended flanking regions using a deep learning method
>> https://www.biorxiv.org/content/10.1101/2023.10.22.563383v1
Deep DNAshape overcomes the limitation of DNAshape, particularly its reliance on the query table search key. This advancement is pivotal, given that the limitation was only caused by the available amount of data.
Deep DNAshape enhances the capability to discern how the shape at the center of a pentamer region is influenced by its extended flanking regions, providing a model that offers a more accurate representation of DNA.
Deep DNAshape can process a given DNA sequence as a string of characters (A, C, G and T) and predict any specific DNA shape for each nucleotide position of a sequence. Deep DNAshape predicts DNA shape and shape fluctuations considering extended flanking influences without biases.

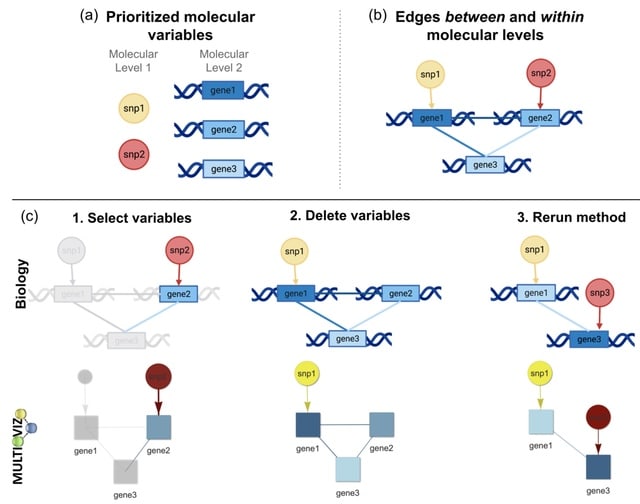
□ NetREm: Network Regression Embeddings reveal cell-type transcription factor coordination for gene regulation
>> https://www.biorxiv.org/content/10.1101/2023.10.25.563769v1
NetREm incorporates information from prior biological networks to improve predictions and identify complex relationships among predictors (e.g. TF-TF coordination: direct/indirect interactions among TFs).
NetREm can highlight important nodes and edges in the network, reveal novel regularized embeddings for genes. NetREm employs Singular Value Decomposition (SVD) to create new latent space gene embeddings, which are then used in a Lasso regression model to predict TG expression.

□ eaDCA: Towards Parsimonious Generative Modeling of RNA Families
>> https://www.biorxiv.org/content/10.1101/2023.10.19.562525v1
eaDCA (Edge Activation Direct Coupling Analysis) is based on an empty coupling network. It then systematically constructs a non-trivial network from scratch, rather than starting with a fully connected network and subsequently simplifying it.
eaDCA operates more swiftly than starting with a fully connected model, leading to generative Potts models. By employing analytical likelihood maximization, it allows to easily track normalized sequence probabilities and estimate entropies throughout the network-building process.

□ BTR: A Bioinformatics Tool Recommendation System
>> https://www.biorxiv.org/content/10.1101/2023.10.13.562252v1
Bioinformatics Tool Recommendation system (BTR) models workflow construction as a session-based recommendation problem and leverage emergent graph neural network technologies to enable a workflow graph representation that captures extensive structural context.
BTR represents the workflow as a directed graph. A variant of the system is constrained to employ linear sequence representations for the purpose of comparison with other methods.
BTR takes the input of Input Query in the format of a sequence: Each tool instance is encoded by an initial embedding layer; The initial embeddings continue to a Gated Graph Neural Network to learn contextural features from neighboring nodes using full workflow graph.
An attention mechanism aggregates the latent graph node embeddings into a full workflow representation, which is concatenated with the representation of the last tool and transformed to yield the final workflow representation vector.

□ DPAMSA: Multiple sequence alignment based on deep reinforcement learning with self-attention and positional encoding
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad636/7323576
DPAMSA (Deep reinforcement learning with Positional encoding and self-Attention for MSA) is based on deep reinforcement learning (DRL). DPAMSA combines natural language processing technology and deep reinforcement learning in MSA.
DPAMSA is mainly based on progressive column alignment, and the sub-alignment of each column is calculated step by step Then all sub-alignments are spliced into a complete alignment.
DPAMSA particularly inserts a gap according to the current sequence state Deep Q Network (DQN) is the deep reinforcement learning model. The model's Q network is divided into positional encoding, self-attention, and multi-layer perceptron.

□ Derived ∞-categories as exact completions
>> https://arxiv.org/abs/2310.12925
A finitely complete ∞-category is exact and additive if and only if it is prestable, extending a classical characterization of abelian categories.
In the ∞-categorical setting, the connection between ∞-topoi, finitary Grothendieck topologies, and coherent ∞-topoi was studied, where it is proven that small hypercomplete ∞-topoi are in correspondence with hypercomplete coherent and locally coherent ∞-topoi.

□ Cyclone: Open-source package for simulation and analysis of finite dynamical systems
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad634/7323572
While there are software packages that analyze Boolean, ternary, or other multi-state models, none compute the complete state space of function-based models over any finite set.
Cyclone simulates the complete state space for an input finite dynamical system and finds all attractors (steady states and limit cycles). Cyclone takes as input functions over any finite set and outputs the entire state space or single trajectories.

□ MultiXrank: Random Walk with Restart on multilayer networks: from node prioritisation to supervised link prediction and beyond
>> https://www.biorxiv.org/content/10.1101/2023.10.18.562848v1
MultiXrank, a Random Walk with Restart algorithm able to explore generic multilayer networks. They define a generic multilayer network as a multilayer network composed of any number and combination of multiplex and monoplex networks connected by bipartite interaction networks.
In this multilayer framework, all the networks can also be weighted and/or directed. MultiXrank outputs scores representing a measure of proximity between the seed(s) and all the nodes of the multilayer network. MultiXrank scores can be used to compute diffusion profiles.

□ LexicHash: Sequence Similarity Estimation via Lexicographic Comparison of Hashes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad652/7329717
LexicHash, a new approach to pairwise sequence similarity estimation that combines the sketching strategy of MinHash with a lexicographic-based hashing scheme.
LexicHash is similar to MinHash in that distinct hash functions are used to create sketches of a sequence by storing the vector of minimum hash values over all k-mers in the sequence.
However, the k-value used in LexicHash actually corresponds to a maximum match length Kmax, and the hashing scheme maintains the ability to capture any match-length below the chosen Kmax.
LexicHash can identify variable-length substring matches between reads from their sketches. The sketches are also constructed in such a way that, to compare sketches, we can traverse the sketches position-by-position, as with the MinHash sketch.

□ gtfsort: a tool to efficiently sort GTF files
>> https://www.biorxiv.org/content/10.1101/2023.10.21.563454v1
gtfsort, a sorting tool that utilizes a lexicographically-based index ordering algorithm. gtfsort not only outperforms similar tools such as GFF3sort or AGAT but also provides a more natural, ordered, and user-friendly perspective on GTF structure.
gtfsort utilizes multiple layers to efficiently write transcript blocks. an outer layer for the highest-level hierarchy, an inner layer for lower-level hierarchies, and a transcript-mapper layer responsible for managing isoforms and their associated features for a given gene.
Each line in the GTF file is parsed and grouped according to its feature, aligning with the specific layer-dependent data flow, including genes, transcripts, and lower-level hierarchies.

□ Back to sequences: find the origin of kmers
>> https://www.biorxiv.org/content/10.1101/2023.10.26.564040v1
back to sequences is dedicated to extracting from a set of sequences, those that contain some of the k-mers given as input and counting the number of occurrences of each of such k-mers. The k-mers can be considered in their original, reverse-complemented, or canonical form.
back to sequences uses the native rust data structures (HashMap) to index and query k-mers. Sequence filtration is based on the minimal and maximal percent of k-mers shared with the indexed set.
On the GenOuest node, back_to_sequences enabled to retrieve all reads that contain at least one of the indexed k-mers in 5m17 with negligible RAM usage of 45MB.
They ran back to_sequences on the full read set, composed of ~ 26.3 billion k-mers, and 381 million reads, again for searching the 69 k-mers contained in its first read. This operation took 20m11.

□ Bioinfo-Bench: A Simple Benchmark Framework for LLM Bioinformatics Skills Evaluation
>> https://www.biorxiv.org/content/10.1101/2023.10.18.563023v1
BIOINFO-BENCH, the bioinformatics evaluation suite to thoroughly assess LLMs' advanced knowledge and problem solving abilities in a bioinformatics scenario. Conducting experiments to evaluate the state-of-the-art LLMs including ChatGPT, Llama, and Galactica on BIOINFO-BENCH.
These LLMs excel in knowledge acquisition, drawing heavily upon their training data for retention. However, their proficiency in addressing practical professional queries and conducting nuanced knowledge inference remains constrained.

□ Scalable genetic screening for regulatory circuits using compressed Perturb-seq
>> https://www.nature.com/articles/s41587-023-01964-9
An alternative approach to greatly increase the efficiency and power of Perturb-seq for both single and combinatorial perturbation screens, inspired by theoretical results from compressed sensing that apply to the sparse and modular nature of regulatory circuits in cells.
To elaborate, perturbation effects tend to be ‘sparse’, in that most perturbations affect only a small number of genes or co-regulated gene programs.
In this scenario, we can measure a much smaller number of random combinations of perturbations and accurately learn the effects of individual perturbations from the composite samples using sparsity-promoting algorithms.

□ PhyloES: An evolution strategy approach for the Balanced Minimum Evolution Problem
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad660/7331089
PhyloES, a novel heuristic that defines the new reference in approximating the optimal solutions to the Balanced Minimum Evolution Problem (BMEP). PhyloES proposes a possible way around this problem that consists in making nondeterministic the search in the solution space.
PhyloES first generates a new set of solutions to the problem by using local search strategies similar to those implemented in FastME. Subsequently, PhyloES stochastically recombines the new phylogenies so obtained by means of the so-called ES operator.
The two phases, the iteration of the local search and the recombination, allow spanning the whole solution space to the BMEP by enabling the potential convergence to the optimum on a sufficiently long period.

□ SeQual-Stream: approaching stream processing to quality control of NGS datasets
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05530-7
SeQual-Stream relies on the Apache Spark framework and the Hadoop Distributed File System (HDFS) to fully exploit the stream paradigm and accelerate the preprocessing of large datasets as they are being downloaded and/or copied to HDFS.
These operations are grouped into three different categories depending on the functionality they provide: (1) single filters, responsible for discarding input sequences that do not meet a certain criteria (sequence length), evaluating each sequence independently of the others;
(2) trimmers, operations that trim certain sequence bases at the beginning or end; and (3) formatters, operations to change the format of the input dataset (DNA to RNA). SeQual-Stream can receive as input single- or paired-end datasets, supporting FASTQ and FASTA formats.

□ cubeVB: Variational Bayesian Phylogenies through Matrix Representation of Tree Space
>> https://www.biorxiv.org/content/10.1101/2023.10.19.563180v1
Using a symmetric matrix with dimension equal to the number of taxa and apply a hierarchical clustering algorithm like the single link clustering algorithm to obtain a tree with internal node heights specified by the values of the matrix.
The entries in the matrix form a Euclidian space, however there are many ways to represent the same tree, so the transformation is not bijective.
By restricting ourselves to the 1-off-diagonal entries of the matrix (and leave the rest at infinity) the transformation becomes a bijection, but cannot represent all possible trees any more.
cubeVB captures the most interesting part of posterior tree space using this approach. cubeVB, a variational Bayesian algorithm based on MCMC and show through well calibrated simulation study that it is possible to recover parameters of interest like tree height and length.

□ LAVASET: Latent Variable Stochastic Ensemble of Trees. A novel ensemble method for correlated datasets
>> https://www.biorxiv.org/content/10.1101/2023.10.20.563223v1
LAVASET derives latent variables based on the distance characteristics of each feature and thereby incorporates the correlation factor in the splitting step. Hence, it inherently groups correlated features and ensures similar importance assignment for these.
LAVASET addresses a major limitation in the interpretation of feature importance of Random Forests when the data are collinear, such as is the case for spectroscopic and imaging data. LAVASET can perform on different types of omics data, from 1D to 3D.

□ ULTRA: Towards Foundation Models for Knowledge Graph Reasoning
>> https://arxiv.org/abs/2310.04562
ULTRA, a method for unified, learnable, and transferable Knowledge Graph (KG) representations that leverages the invariance of the relational structure and employs relative relation representations on top of this structure for parameterizing any unseen relation.
ULTRA constructs a graph of relations (where each node is a relation from the original graph) capturing their interactions. ULTRA obtains a unique relative representation of each relation. It enables zero-shot generalization to any other KG of any size and any relation.

□ PerFSeeB: designing long high-weight single spaced seeds for full sensitivity alignment with a given number of mismatches
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05517-4
The PerFSeeB is based on designing periodic blocks. When several mismatches are set, resulting spaced seeds are guaranteed to find all positions within a reference sequence. Each periodic seed consists of an integer number of periodic blocks and a “remainder”.
Those blocks can be used to generate spaced seeds required for any given length of reads. The best periodic seeds are seeds of maximum possible weight since this helps us to reduce the number of candidate positions when we try to align reads to the reference sequence.

□ Benchmarking algorithms for joint integration of unpaired and paired single-cell RNA-seq and ATAC-seq data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03073-x
The incorporation of multiome data improves the cell type annotation accuracy of scRNA-seq and snATAC-seq data when there are a sufficient number of cells in the multiome data to reveal cell type identities.
When generating a multiome dataset, the number of cells is more important than sequencing depth for cell type annotation. Seurat v4 is the best at integrating scRNA-seq, snATAC-seq, and multiome data even in the presence of complex batch effects.

□ OMEinfo: Global Geographic Metadata for -omics Experiments
>> https://www.biorxiv.org/content/10.1101/2023.10.23.563576v1
OMEinfo leverages open data sources such as the Global Human Settlement Layer, Köppen-Geiger climate classification models, and Open-Data Inventory for Anthropogenic Carbon dioxide, to ensure metadata accuracy and provenance.
OMEinfo's Dash application enables users to visualise their sample metadata on an interactive map and to investigate the spatial distribution of metadata features, which is complemented by data visualisation to analyse patterns and trends in the geographical data.

□ MGA-seq: robust identification of extrachromosomal DNA and genetic variants using multiple genetic abnormality sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03081-x
MGA-Seq (multiple genetic abnormality sequencing) simultaneously detect structural variation, copy number variation, single-nucleotide polymorphism, homogeneously staining regions, and extrachromosomal DNA (ecDNA) from a single tube.
MGA-Seq directly sequences proximity-ligated genomic fragments, yielding a dataset with concurrent genome three-dimensional and whole-genome sequencing information, enabling approximate localization of genomic structural variations and facilitating breakpoint identification.

□ Open MoA: Revealing the Mechanism of Action (MoA) based on Network Topology and Hierarchy
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad666/7334463
Open MoA computes confidence scores to edges that represent connections between genes/proteins in the integrated network. The interactions showing the highest confidence score could indicate potential drug targets and infer the underlying molecular MoAs.
Open MoA reveasl the MoA of a repositioned drug (JNK-IN-5A) that modulates the PKLR expression in HepG2 cells and found STAT1 is the key transcription factor.
With the transcriptomic data, by inputting the known starting point and endpoints, Open MoA is able to give out the significant confidence score for each interaction in the context-specific subnetworks, thus leading to the identification of the most possible pathway.

□ ORI-Explorer: A unified cell-specific tool for origin of replication sites prediction by feature fusion
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad664/7334464
ORI-Explorer, a unique AI-based technique that combines multiple feature engi- neering techniques to train CatBoost Classifier for recognizing ORIs from four distinct eukaryotic species.
ORI-Explorer was created by utilizing a unique combination of three traditional feature-encoding techniques and a feature set obtained from a deep-learning neural network model.
ORI-Explorer uses 4 different feature descriptors, where one is extracted using the distinctive neural network architecture while the other 3 are composition k-spaced nucleic acid pairs, Parallel Correlation Pseudo Dinucleotide Composition and Dinucleotide-based Cross Covariance.
While these features are concatenated and given to SHapley Additive exPlanation (SHAP) to select the most important features that are further used by CatBoost to predict the ORI regions.

□ High-fidelity (repeat) consensus sequences from short reads using combined read clustering and assembly
>> https://www.biorxiv.org/content/10.1101/2023.10.26.564123v1
The presented repeat assembly workflow uses clustering and assembly tools to create informed consensus sequences from repeats to answer a wide variety of questions.
They use the term "informed consensus" to suggest that the derived sequences are not mere averages or sequence profiles, but that they have been carefully constructed using relevant data and analysis.

□ DENIS: Uncovering uncharacterized binding of transcription factors from ATAC-seq footprinting data
>> https://www.biorxiv.org/content/10.1101/2023.10.26.563982v1
DENIS (DE Novo motlf diScovery) that i) isolates UBM events from ATAC-seq data, ii) performs de novo motif generation, iii) calculates information content, motif novelty and quality parameters, and iv) characterizes de novo motifs through open chromatin enrichment analysis.
DENIS is designed to robustly explore DNA binding events on a global scale, to compare ATAC-seq datasets from one or multiple conditions, and is suitable to be applied to any organism.
DENIS merges very similar motifs found in multiple iterations and continues with the consensus motif. DENIS generated a total of 141 motifs over 26 iterations, which were finally merged to 30 unique motifs.

□ CHESS 3: an improved, comprehensive catalog of human genes and transcripts based on large-scale expression data, phylogenetic analysis, and protein structure
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03088-4
CHESS 3 takes a stricter approach to including genes and transcripts than other human gene catalogs. CHESS 3 represents an improved human gene catalog based on nearly 10,000 RNA-seq experiments across 54 body sites.
CHESS 3 contains 41,356 genes, including 19,839 protein-coding genes and 158,377 transcripts, with 14,863 protein-coding transcripts not in other catalogs. It includes all MANE transcripts and at least one transcript for most RefSeq and GENCODE genes.