









□ math, astronomy and biology.
選択圧は言語においても作用する。言語に作用するものは行動を規定するのだから、我々は忌避すべき可能性の一つ一つを、己の意思だけで剪定できるわけではないことを知っている。法の抑止とは、即ち選択圧の制御である。
□ Discrete dynamical model determining the biodiversity & stability at different LVs of organization of living matter:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/11/161687.full.pdf
The stability of biological systems provided by the mechanisms of homeostasis is a sort of dynamic equilibrium, which assumes fluctuations in the values of system’s parameters within in a certain cycle.
□ DEEP LEARNING DEAD LANGUAGES:
>> http://www.3quarksdaily.com/3quarksdaily/2017/01/deep-learning-dead-languages.html
Among the family of Sámi languages (of the indigenous groups of northern Norway, Sweden, Finland and Russia), several of the languages are already extinct or with very few and old speakers left, but efforts are being made to help revive some of them.
□ Aether: Leveraging Linear Programming for Optimal Cloud Computing In Genomics:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/13/162883.full.pdf
Aether’s replica nodes specified by the linear programming result are launched and placed under the control of a primary node, which assigns batch processing jobs over Transmission Control Protocol, monitors for any failures, gathers all logs, sends all results to a specified cloud storage location, and terminates all compute nodes once processing is complete. using Aether, 30 instances 1TB RAM/instance cost is $0.3/assembly, $471.60 for 1572 assemblies in 13 hours total.
□ HECIL: A Hybrid Error Correction Algorithm for Long Reads with Iterative Learning:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/13/162917.full.pdf
a convex combination of the objectives as in eq. rather than formulating a multi-objective optimization problem & Pareto-optimal solutions. HECIL core algorithm could also be seamlessly integrated with other error correction algorithms that leverage short read alignments.

□ A phase transition induces chaos in a predator-prey ecosystem with a dynamic fitness landscape:
>> http://plos.io/2tyN4Hj
Despite the large fluctuations in the maximum value of the local Lyapunov exponents, the largest global Lyapunov exponent is only barely larger than zero, λmax≈0.003. Similar behavior has been reported a real-world ecosystem consisting of competing species in a rocky intertidal environment, in which a small global Lyapunov exponent paired with a fluctuating largest local Lyapunov exponent was taken to suggest that the ecosystem had adapted to “the edge of chaos.”

□ DNA sequence+shape kernel enables alignment-free modeling of transcription factor binding:
>> https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btx336
the RBF kernel might provide an alternate solution for the high-dimensionality issues in a shape-augmented models. The RBF kernel embeds the data into an infinite dimensional feature space & efficient mapping to a high-dimensional, implicit feature space.

□ Complexity-Entropy maps as a tool for the characterization of the clinical electrophysiological evolution:
>> http://www.biorxiv.org/content/early/2017/07/16/164236
analyzing a sequence from a completely deterministic point of view, CLZ(Sn) also contains a notion of information in a statistical sense. This make it emerge a relation between the Lempel–Ziv complexity and the Shannon entropy.
□ Nanofilt using albacore sequencing_summary for quality filtering.
>> https://gigabaseorgigabyte.wordpress.com/2017/07/15/nanofilt-using-albacore-sequencing_summary-for-quality-filtering/
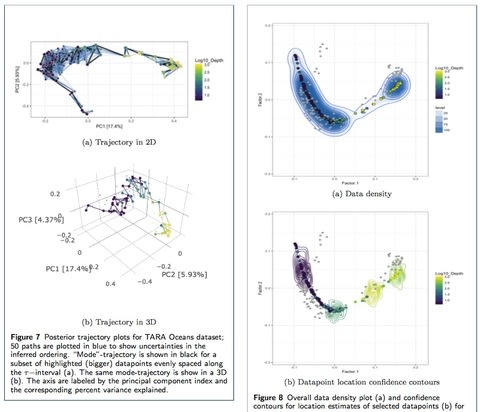
□ Bayesian Unidimensional Scaling for visualizing uncertainty in high dimensional data with latent ordering of observations
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/18/163915.full.pdf
By statistically modeling dissimilarities and applying a DiSTATIS registration method to their posterior samples, and able to incorporate visualizations of uncertainties in the estimated data trajectory across different regions using confidence contours for individual data points.
□ ShinyGPAS: Interactive genomic prediction accuracy simulator based on deterministic formulas:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/17/164772.full.pdf
Each of deterministic formula captures a different aspect of the genotype-phenotype map in the context of genomic prediction accuracy. Thus, navigating interactively visualized deterministic formulas may highlight the common patterns as well as differences among them.
□ On pairwise alignment with dynamic programming:
>> https://figshare.com/articles/Notes_on_pairwise_alignment_with_dynamic_programming/5223973
SIMD has been used to speed up DP-based pairwise alignment. There are two general classes of algorithms: inter-sequence and intra-sequence. Inter-sequence algorithms align multiple pairs of sequences at the same time. It is conceptually easier to implement and faster to run but it is tricky to use with other alignment routines. Intra-sequence algorithms align one sequence at a time. There are several ways to implement intra-sequence pairwise alignment, depending on how to organize multiple data into one vector.

□ druGAN: generative adversarial autoencoder for de-novo generation of new molecules with desired properties in silico
>> http://pubs.acs.org/doi/pdf/10.1021/acs.molpharmaceut.7b00346
an advanced AAE model for molecular feature extraction problems and proved its superiority to VAE in terms of adjustability in generating molecular fingerprints; capacity of processing huge molecular datasets; and efficiency in unsupervised pretraining for the regression model.
□ Language evolution to revolution: developing finite communication system with many words to infinite modern language
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/20/166520.full.pdf
a “Romulus and Remus” hypothesis of language acquisition and proposed that recursive language elements such as spatial prepositions were acquired synergistically with mental synthesis 100,000 years ago in one revolutionary jump towards modern infinite language.
□ Hybrid assembly with long and short reads improves discovery of gene family expansions:
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3927-8
Alpaca (ALLPATHS and Celera Assembler) that exploits existing tools to assemble Illumina short-insert paired-end short reads (SIPE), Illumina long-insert paired-end short reads (LIPE), and PacBio unpaired long reads. long-read data in order to make the pipeline a practical tool for non-model systems and for surveys of intraspecific structural variation.
□ ISMB/ECCB 2017 Conference in Prague (July 21-25.)
>> https://www.iscb.org/ismbeccb2017
□ BOSC 2017 the Open Bioinformatics Foundation, (July 22-23)
>>https://www.open-bio.org/wiki/BOSC_2017
□ Rabix Executor: an open-source executor recomputability and interoperability of workflow descriptions:
>> https://github.com/rabix/bunny #BOSC2017
Common Workflow Language bindings that integrates the executor with backends supporting GA4GH TEST, LSF, Slurm. String persistence = configuration.getString("engine. store", "IN_MEMORY");
if (persistence.equals("POSTGRES")) {
□ Towards a scientific blockchain framework for reproducible data analysis:
>> https://arxiv.org/pdf/1707.06552.pdf
The proponent deposits a pre-determined amount of probos coins to start a smart-contract-like mechanism which broadcasts her request to the PROBO network, where other researchers/labs (the verifiers) will evaluate the quality of the study and verify its reproducibility.
□ Cross-linking BioThings APIs through JSON-LD to facilitate knowledge exploration:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/23/167353.full.pdf
quantify the extent of differences in annotation of 424 million variants from multiple variant annotation DB, incl ClinVar, dbSNP & dbNSFP. a mechanism for expressing in a structured format the semantic nature between concepts and the provenance of such relationships would expand the richness of possible queries in a JSON-LD ecosystem.

□ DEEPred: Multi-task Deep Neural Networks in Automated Protein Function Prediction∗:
>> https://arxiv.org/pdf/1705.04802.pdf
deep learning can be employed to significantly improve the performance of prediction (F-score > 0.75) for hard to predict GO categories such as the biological process and the cellular component, especially when the training set sizes are sufficiently large (¿ 500 proteins).
□ A Fast Approximate Algorithm for Mapping Long Reads to Large Reference Databases:
>> https://link.springer.com/chapter/10.1007/978-3-319-56970-3_5
a fast approximate read mapping algorithm based on minimizers w/ novel MinHash identity estimation to achieve both scalability & precision. For mapping human PacBio reads to the hg38 reference, this method is 290x faster than BWA-MEM with a lower memory and recall rate of 96%.

□ GridProd: Grid-based computational methods for the design of constraint-based parsimonious chemical reaction networks
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/24/166777.full.pdf
GridProd divides the solution space of FBA into P −2 small grids, and conducts LP twice for each grid. The area size of each grid is (P × T M GR) × (P × T M P R). TMPR stands for theoretical maximum production rate.
□ VALOR: Discovery of large genomic inversions using long range information such as 10X linked-read sequencing:
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-3444-1
the paired-end reads from a total of 288 pools to the reference genome using BWA-MEM. Average fragment length of the paired-end reads was ~450 bp, with a standard deviation of ~98 bp. Using Valor, we reconstructed the clone locations, which showed an average clone length of ~140 Kbp and a standard deviation of ~40 Kbp. The Chicago method from Dovetail Genomics, TruSeq Synthetic Long Reads, and the CPT-Seq are other candidates for further VALOR development.
□ The language of geometry: Fast comprehension of geometrical primitives and rules:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005273
Even for simple languages, Kolmogorov complexity is often difficult to compute in practice, because it involves examining, for each sequence, all the programs that compute it, a search that typically grows exponentially with the size of the sequence. Different methods have been developed to Kolmogorov complexity. One is to approximate it using standard file compressor such as Lempel-Ziv.

□ PRESAGE: a hybrid framework of hardware & multiple crypto protocols protects genomic data against malicious attacks:
>> https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-017-0281-2
For a VCF file w/ 200K records, the PRESAGE securely processes a query w/n 0.05s, which includes file loading, unsealing and query matching. Compared with state-of-the-art HME solution, PRESAGE framework shows at least 120X performance gain.

□ DiGeST: Distributed Computing for Scalable Gene and Variant Ranking with Hadoop/Spark:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/27/168633.full.pdf
A compelling advantage of the frameworks is that most of the complexity related to the distributed infrastructure is abstracted by the HDFS storage system and the MapReduce programming model.
□ Shapes Constraint Language (SHACL):
>> https://www.w3.org/TR/2017/REC-shacl-20170720/
The SHACL Core language defines two types of shapes:
1. shapes about the focus node itself, called node shapes
2. shapes about the values of a particular property or path for the focus node, called property shapes.
sh:deactivated : xsd:boolean
sh:message : xsd:string or rdf:langString
sh:severity : sh:Severity

□ De novo Clustering Nanopore Long Reads of Transcriptomics Data by Gene:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/30/170035.full.pdf
CARNAC is parameter-free, needs no pre-processing of the data and performs its clustering task while being very precise and thus reliable. a clustering technique that aims at finding cuts in graphs of read similarities in order to make a partition that separates subgraphs gathering similar reads, while taking account of the quasi-clique nature of these subgraphs.

□ Learning causal biological networks with generalized Mendelian randomization:
>> http://www.biorxiv.org/content/biorxiv/early/2017/08/01/171348.full.pdf
This method analyzes a data matrix with each row being an individual, and each column a genetic variant or a molecular phenotype. Like most causal graph learning methods, a key assumption behind MRPC is that there are no hidden nodes are connected to the observed nodes.

□ TRuML: A Translator for Rule-Based Modeling Languages:
>> http://www.biorxiv.org/content/biorxiv/early/2017/08/01/171306.full.pdf
TRuML accommodates the languages’ complexities and produces a semantically equivalent model in the alternate language of the input model. Kappa language and the BioNetGen language encode types of molecules and their interactions as human- & machine-readable plain text as a name and a list of sites that can interact with other molecules’ sites or occupy one of a finite number of predefined states.

□ Linear Assembly of a Human Y Centromere using Nanopore Long Reads:
>> http://www.biorxiv.org/content/biorxiv/early/2017/07/31/170373.full.pdf
a long-read strategy to advance sequence characterization of tandemly repeated satellite DNAs. and focused on the haploid satellite array that spans the Y centromere (DYZ3) as it is particularly suitable for assembly due to its tractable array size, well-characterized higher order repeats (HORs) structure and previous physical mapping data.

□ Bayesian Dirichlet Bayesian Network Scores and the Maximum Entropy Principle:
>> https://arxiv.org/pdf/1708.00689.pdf
the Bayesian Dirichlet equivalent uniform score may select models in a way that violates maximum entropy principle. In contrast, the BDs score proposed in Scutari does not, even though it converges to BDeu asymptotically. Maximum Entropy directly incorporates the information encoded in the hyper-prior w/o relying on large samples to link the empirical entropy.

□ SuperTranscripts: a data driven reference for analysis and visualisation of transcriptomes:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1284-1
superTranscripts provide a convenient way of looking at the coverage & expression levels of long-read data such as PacBio or Nanopore data. Lace works by building a splice graph for each gene, then topologically sorting the graph using Kahn’s algorithm. Lace annotates each superTranscript with blocks either by using the graph structure itself or alternatively using a script called Mobius.
□ DeepATAC: A deep-learning method to predict regulatory factor binding activity from ATAC-seq signals:
>> http://www.biorxiv.org/content/biorxiv/early/2017/08/06/172767.full.pdf
DeepATAC integration provides a 40% average performance improvement over DeepSEA.
doublearm.compile(optimizer=optimizers.SGD(lr=1e-4, momentum=0.9),
loss='binary_crossentropy',
metrics=['accuracy'])