□ A divide-and-conquer algorithm for large-scale de novo transcriptome assembly through combining small assemblies from existing algorithms:
>>
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4270-9
While the memory requirement can still be high even after applying the divide-and-conquer strategy on memory-intensive algorithms for very large data sets, they are generally more accurate, with Oases returning more and longer transcripts and Trinity returning more transcripts with low expression levels and with less translocations. Among the memory-efficient algorithms, SOAPdenovo-Trans returns transcripts with less translocations while Trans-ABySS returns more and longer transcripts with higher specificity.
□ SemEHR: A General-purpose Semantic Search System to Surface Semantic Data from Clinical Notes for Tailored Care, Trial Recruitment and Clinical Research:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/18/235622.full.pdf
□ Analyzing Complete Genomics (GNOM) and Tabula Rasa HealthCare (TRHC)
>>
https://ledgergazette.com/2017/12/18/financial-analysis-tabula-rasa-healthcare-trhc-complete-genomics-gnom-2.html

□ Your ancestors lived all over the world, but relatively few of them were your genetic ancestors (does that matter?)
>>
https://gcbias.org/2017/12/19/1628/
□MEBoost: Mixing Estimators with Boosting for Imbalanced Data Classification:
>>
https://arxiv.org/pdf/1712.06658.pdf
MEBoost mixes two different weak learners with boosting to improve the performance on imbalanced datasets. MEBoost is an alternative to the existing techniques such as SMOTEBoost, RUSBoost, Adaboost, etc.
□ Some great plots showing flow cell trends utilising data from ~800 nanopore runs at Genoscope in this presentation by @J_M_Aury
>>
http://www.genoscope.cns.fr/externe/rna_workshop/slides/JeanMarc_Aury.pdf
6 MinION devices >800 flowcells; >50 different organisms; ~700Gb of ONT reads ; DNA and RNA samples.
Based on Lexogen ́s unique Cap-Dependent Linker Ligation (CDLL) and long reverse transcription (long RT) technology, it is highly selective for full- length RNA molecules that are both capped and polyadenylated.
□ The Norwegian government's New national strategy for acces to, and sharing and of, research data is out!
>>
https://www.regjeringen.no/no/dokumenter/nasjonal-strategi-for-tilgjengeliggjoring-og-deling-av-forskningsdata/id2582412/
□ FinnGen project announced: 10% of Finns profiled and linked to decades of nation wide EHR data: 9 biobanks, 7 pharma partners, public-private partnership, terrific opportunities to develop precision medicine. @FIMM_UH @HiLIFE_helsinki @THLorg
□ bioconda singularity containers
>>
https://depot.galaxyproject.org/singularity
□ Nanopore DNA Sequencing and Genome Assembly on the International Space Station
>>
https://www.nature.com/articles/s41598-017-18364-0

□ bioSyntax: Syntax Highlighting For Computational Biology:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/20/235820.full.pdf
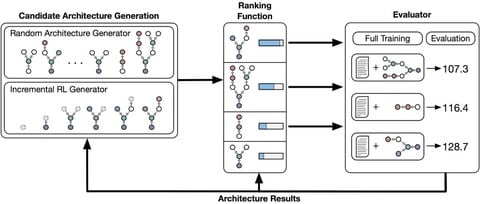
□ A domain specific language for automated rnn architecture search:
>>
https://einstein.ai/research/domain-specific-language-for-automated-rnn-architecture-search
define a DSL for constructing RNNs and then filter candidates by estimating their performance using a TreeLSTM ranking function. A generator produces candidate architectures by iteratively sampling the next node (either randomly or using an RL agent trained with REINFORCE).

□ VaDiR: an integrated approach to Variant Detection in RNA:
>>
https://watermark.silverchair.com/gix122.pdf
VaDiR integrates three variant callers, namely: SNPiR, RVBoost and MuTect2. The combination of all three methods, which they called Tier1 variants, produced the highest precision with true positive mutations from RNA-seq that could be validated at the DNA level. They also found that the integration of Tier1 variants with those called by MuTect2 and SNPiR produced the highest recall with acceptable precision.
□ Tetrapods on the EDGE: Overcoming data limitations to identify phylogenetic conservation priorities:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/21/232991.full.pdf
The EDGE metric, which prioritises species based on their Evolutionary Distinctiveness (ED) and Global Endangerment (GE), relies on adequate phylogenetic and extinction risk data to generate meaningful priorities for conservation. To overcome paucity of genetic data, many phylogenies are now constructed using taxonomic information and constraints to infer phylogenetic relationships for species lacking available genetic data.

□ Tellurium Notebooks - An Environment for Dynamical Model Development, Reproducibility, and Reuse:
>>
https://www.biorxiv.org/content/early/2017/12/23/239004
Tellurium, a Python–based Jupyter–like environment, is designed to seamlessly inter-operate with these community standards by automating conversion between COMBINE standards formulations and corresponding in–line, human–readable representations. Tellurium supports embedding human–readable representations of SBML and SED–ML directly in cells. These cells can be exported as COMBINE archives which are readable by other tools, refer to human–readable representation as inline OMEX (after Open Modeling and EXchange).
□ An accurate and rapid continuous wavelet dynamic time warping algorithm for unbalanced global mapping in nanopore sequencing:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/23/238857.full.pdf
a novel dynamic time warping algorithm, cwDTW, based on continuous wavelet trans- form (CWT), to cope with the unbalanced global mapping between two ultra-long signal sequences. the algorithm has an approximate O(N) time- and space-complexity, where N is the length of the longer sequence, and substantially advances previous methods in terms of the mapping accuracy.
□ DeepSimulator: a deep simulator for Nanopore sequencing:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/22/238683.full.pdf
DeepSimulator using a novel deep learning strategy BiLSTM-extended Deep Canonical Time Warping (BDCTW), which combines bi-directional long short-term memory (Bi-LSTM) with deep canonical time warping (DCTW) to solve the scale difference issue. deep canonical time warping architecture w/ two DNNs, one for the input nucleotide sequence (one-hot encoding for each nucleotide thus the feature dimension is four) and the other for the observed electrical current measurements (denoted as raw signals w/ feature dimension one).

□ Mirnovo: genome-free prediction of microRNAs from small RNA sequencing data and single-cells using decision forests:
>>
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5716205/pdf/gkx836.pdf
this method has been validated using large-scale datasets and canonical biogenesis mutant datasets that elucidate potential novel miRNA biogenesis pathways, based on their dependency on different types of RNaseIII enzymes.

□
AlbertVilella:
Updated S1, S2 and S4 @illumina NovaSeq flowcell pricing as well as Oxford Nanopore pricing and throughput (from @nanopore site when available) and BGISEQ-500 pricing.
□ ClassificaIO: machine learning for classification graphical user interface
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/28/240184.full.pdf
ClassificaIO is an open-source Python graphical user interface for supervised machine learning classification for the scikit-learn module. ClassificaIO is a Python library with the following external dependencies: nltk ≥ 3.2.5, Tcl/Tk ≥ 8.6.7, Pillow ≥ 4.3, pandas ≥ 0.21, numpy ≥ 1.13, scikit-learn ≥ 0.19.1. and they recommend using the Spyder integrated development environment (IDE) in Anaconda Navigator.
□ Whisper: Read sorting allows robust mapping of sequencing data:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/28/240358.full.pdf
Whisper excels at large NGS read collections, in particular Illumina reads with typical WGS coverage. The experiments with real data indicate that solution works in about 15% of the time needed by the well-known Bowtie2 and BWA-MEM tools at a comparable accuracy. Although Whisper essentially handles up to k errors, some matches with more (up to 3k by default) Levenshtein errors are also detected.
□ The Functional False Discovery Rate with Applications to Genomics:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/30/241133.full.pdf
the fFDR methodology to utilize additional information on the prior probability of a null hypothesis being true or the power of the family of test statistics in multiple testing. It employs a functional null proportion of true null hypotheses and a joint density for the p-values and informative variable.
qvalue <- function(p, fdr.level = NULL, pfdr = FALSE, lfdr.out = TRUE, pi0 = NULL, ...) {
# Argument checks
p_in <- qvals_out <- lfdr_out <- p
rm_na <- !is.na(p)
p <- p[rm_na]
if (min(p) < 0 || max(p) > 1) {
stop("p-values not in valid range [0, 1].")
}else if (!is.null(fdr.level) && (fdr.level <= 0 || fdr.level > 1)) {
stop("'fdr.level' must be in (0, 1].")
}

□ DeepGS: Predicting phenotypes from geno- types using Deep Learning:
>>
https://www.biorxiv.org/content/biorxiv/early/2017/12/31/241414.full.pdf
A representative example is the commonly used RR-BLUP model, which assumes that all the marker effects are normally distributed with a small but non-zero variance, and predicts phenotypes from a linear function of genotypic markers. An integrated GS model (I) was constructed using the ensemble learning approach by linearly combining the predictions of DeepGS (D) and RR- BLUP (R), using the formula:
predictl = (WD * predictD + WR * predictR)/(WD + WR)

□ DE-kupl: exhaustive capture of biological variation in RNA-seq data through k-mer decomposition:
>>
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1372-2
1. Indexing: index and count all k-mers (k=31) in the input libraries
2. Filtering and masking: delete k-mers representing potential sequencing errors or perfectly matching reference transcripts
3. Differential expression (DE): select k-mers with significantly different abundances across conditions
4. Extending and annotating: build k-mer contigs and annotate contigs based on sequence alignment.
A key aspect of this protocol that rendered a full k-mer analysis tractable was the application of successive filters for rare k-mers, reference transcripts, and DE, which altogether resulted in a 200-fold reduction in k-mer counts.

□ DECODE-ing sparsity patterns in single-cell RNA-seq:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/01/01/241646.full.pdf
DECODE using a dynamic-programming approach to assign an exact p-value to the optimal neighborhood size and predicts that a primarily inactive network neighborhood is likely to reveal true biological zeros of a cell. if the proposed framework is consistent with function, aiming to be able to predict missing values (fill in the diagonal blocks) without perturbing biological zeros (off-diagonal elements). For non-biological zeros, building a predictive model to impute the missing value using their most informative neighbors and their framework accurately infers gene-gene functional dependencies, pinpoints technical zeros, and biologically-meaningful missing values.
□ DNAnexus Closes $58M Venture Capital Round
>>
https://www.genomeweb.com/business-news/dnanexus-closes-58m-venture-capital-round
DNAnexus, maker of a cloud-based genome informatics and data management platform, has closed a $58 million round of venture capital. New investor Foresite Capital led the round, while Microsoft made a "strategic investment," Mountain View, California-based DNAnexus said today. Previous investors GV, TPG Biotech, WuXi NextCode, Claremont Creek Ventures, and MidCap Financial also took part in the financing.

□ Invitae - The Next Generation Of Medicine Is Genomics:
>>
https://seekingalpha.com/article/4135706-invitae-next-generation-medicine-genomics
Founder and Executive Chairman ran Genomic Health from a start-up into a public company worth $1 billion. The company has a sinking bottom line and recorded a net loss of $82.9 million for the three quarters ending 2017 which had widened compared to the same time period of $75.4 million in 2016. Revenues rose 189%, gross profits increased $5.84 million, volume was up 158%, and COGS per sample was down 26.7% for the quarter compared to the same quarter of the prior year.
□ the Flye assembler for ONT / PacBio reads (successor of ABruijn)
>>
https://github.com/fenderglass/Flye
It's accurate, scales to the human genome, but most importantly it generates fancy assembly graph ouput. Flye is a de novo assembler for long and noisy reads, such as those produced by PacBio and Oxford Nanopore Technologies. The algorithm uses an A-Bruijn graph to find the overlaps between reads and does not require them to be error-corrected. After the initial assembly, Flye performs an extra repeat classification and analysis step to improve the structural accuracy of the resulting sequence. The package also includes a polisher module, which produces the final assembly of high nucleotide-level quality.

□ A mixed quantum chemistry/machine learning approach for the fast and accurate prediction of biochemical redox potentials and its large-scale application to 315,000 redox reactions:
>>
https://www.biorxiv.org/content/biorxiv/early/2018/01/09/245357.full.pdf
the following combination of quantum model chemistry and kernel/distance function between reactions resulted in the most efficient prediction strategy: the semiempirical method for both geometry optimizations and single point electronic energies, with COSMO implicit solvatio, and a reaction fingerprint obtained by taking the difference between the Morgan fingerprint vectors of products and substrates, with a kernel function that is a mixture of a squared exponential kernel and a noise kernel.
□
mason_lab:
First run on the new @illumina Firefly sequencer looks great! 6M 151x151 PF reads, 1.8GBases, >99.8% accuracy in 16 hours. Santa came with SBS candy in a CMOS wrapper this year.
□ Illumina 2018 Preview I: Firefly:
>>
http://omicsomics.blogspot.com/2018/01/illumina-2018-preview-i-firefly.html
Firefly delivers about 80% as much 2x150 data in a similar amount of time. if your application can ride out the sequence quality issues of Oxford Nanopore, that $30K pricetag would buy and awful lot of MinION flowcells and practiced users can get 2Gbases of long read data in a few hours, including library prep.
□ 36th Annual J.P. Morgan HEALTHCARE CONFERENCE January 8 - 11, 2018
>>
https://www.jpmorgan.com/global/healthcareconference
□
pathogenomenick:
#AGBT18 programme is up!
>>
http://www.agbt.org/gm-agenda/
@Scalene talking about ultra-long reads on nanopore
@thekrachael talking about direct RNA sequencing on nanopore
□ GATK4 ON DNANEXUS
>>
https://blog.dnanexus.com/2018-01-09-gatk4-on-dnanexus/
dxWDL takes a bioinformatics pipeline written in Workflow Description Language and compiles it to an equivalent workflow on the DNAnexus platform. WDL supports complex and recursive data types, which do not have native support. In order to maintain the usability of the UI, map WDL types to the dx equivalent. This works for primitive types (Boolean, Int, String, Float, File), and for single dimensional arrays of primitives.











