□ Accel-Align: A Fast Sequence Mapper and Aligner based on the Seed-Embed-Extend Method
>> https://www.biorxiv.org/content/10.1101/2020.07.20.211888v1.full.pdf
seed–embed–extend (SEE), a new design methodology for developing sequence mappers and aligners. While seed–filter–extend (SFE) focuses on eliminating sub-optimal candidates, SEE focuses instead on identifying optimal candidates.
SEE transforms the read and reference strings from edit distance regime to the Hamming regime by embedding them using a randomized algorithm, and uses Hamming distance over the embedded set to identify optimal candidates.
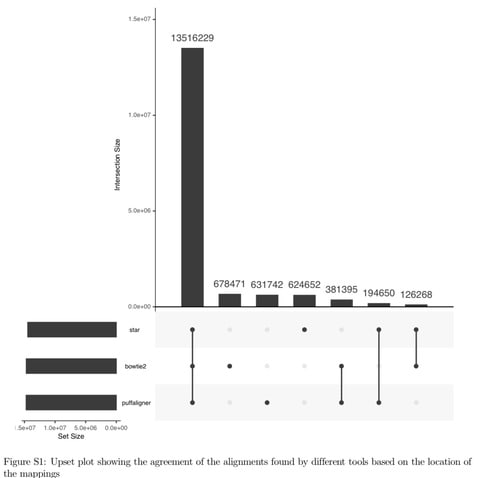
Accel-Align clearly outperforms the other aligners, as it is 9× faster than Bowtie2, 6× faster than BWA-MEM, and 3× faster than Minimap2.
Accel-Align calculates the Hamming distance between each embedded candidate reference and the read, and selects the two best candidates with the lowest Hamming distance. Accel-Align processes each read by first extracting seeds to find candidate locations similar to SFE aligners.
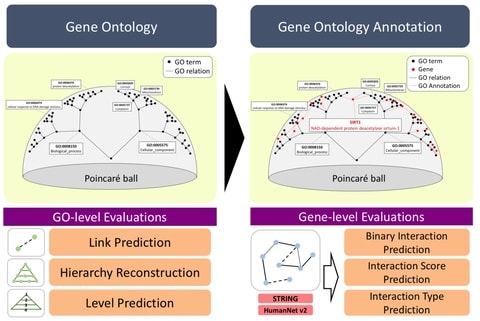
□ HiG2Vec: Hierarchical Representations of Gene Ontology and Genes in the Poincaré Ball
>> https://www.biorxiv.org/content/10.1101/2020.07.14.195750v1.full.pdf
The problem of dimensional decision involves a tradeoff. According to the word embedding that has been studied, low-dimensional space is not expressive enough to capture the entire relation, and high-dimensional space has powerful representation ability but is susceptible to overfitting.
the HiG2Vec embedding on the Poincar ́e ball can limit its application in Euclidean space, but it can be applied to general machine learning or deep learning based applications. HiG2Vec outperformed all other embedding methods on the 1,000-dimensional space.
□ A sparse Bayesian factor model for the construction of gene co-expression networks from single-cell RNA sequencing count data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03707-y
the high resolution of scRNA-seq technology allows researchers the opportunity to estimate “pseudotime” and obtain a temporal ordering of cells. a sparse hierarchical Bayesian factor model to explore the network structure associated with genes.
Latent factors impact the gene expression values for each cell and provide flexibility to account for common features of scRNA-seq: high proportions of zero values, increased cell-to-cell variability, and overdispersion due to abnormally large expression counts.
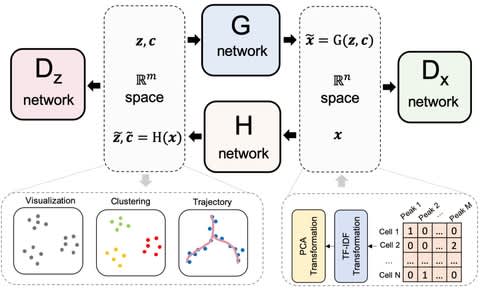
□ scDEC: Simultaneous deep generative modeling and clustering of single cell genomic data
>> https://www.biorxiv.org/content/10.1101/2020.08.17.254730v1.full.pdf
scDEC is built on a pair of generative adversarial networks (GANs), and is capable of learning the latent representation and inferring the cell labels, simultaneously.
scDEC consists of two GAN models, which are utilized for transformations b/w latent space and data space. the way of latent indicator interpolation in the data generation can be further explored, especially in a complicated tree or graph-based trajectory of cell differentiation.

□ uLTRA: a long transcriptomic read aligner
>> https://github.com/ksahlin/ultra
uLTRA is a tool for splice alignment of long transcriptomic reads to a genome, guided by a database of exon annotations. uLTRA takes reads in fast(a/q) and a genome annotation as input and outputs a SAM-file.
uLTRA can be used with either Iso-Seq or ONT reads. It outputs to extra tags describing whether all the splices sites are known and annotated (FSM), new splice combinations (NIC). uLTRA is highly accurate when aligning to small exons.

□ scGNN: a novel graph neural network framework for single-cell RNA-Seq analyses
>> https://www.biorxiv.org/content/10.1101/2020.08.02.233569v1.full.pdf
a multi-modal framework scGNN (single-cell graph neural network), which synergistically determines cell clusters based on a bottom-up integration of detailed pairwise cell-cell relationships and the convergence of predicted clusters.
scGNN utilizes GNN with multi-modal autoencoders to formulate and aggregate cell-cell relationships, providing a hypothesis-free framework. Cell-type-specific regulatory signals are modeled in building a cell graph, equipped with a left-truncated mixture Gaussian (LTMG) model.
□ SpatialDecon: Advances in mixed cell deconvolution enable quantification of cell types in spatially-resolved gene expression data
>> https://www.biorxiv.org/content/10.1101/2020.08.04.235168v1.full.pdf
SpatialDecon obtains cell abundance estimates that are spatially-resolved, granular, and paired with highly multiplexed gene expression data.
The SpatialDecon algorithm was applied to all segments in the dataset using the SafeTME matrix. Log-normal regression has the same theoretical benefits in bulk expression deconvolution.

□ Ratatosk - Hybrid error correction of long reads enables accurate variant calling and assembly
>> https://www.biorxiv.org/content/10.1101/2020.07.15.204925v1.full.pdf
Ratatosk can reduce the raw error rate of Oxford Nanopore reads 6-fold on average with a median error rate as low as 0.28%. Ratatosk corrected data maintain nearly 99% accurate SNP calls and substantially increase indel calls accuracy by up to about 40% compared to the raw data.
Long reads are subsequently anchored on the graph using exact and inexact k-mer matches to find paths corresponding to corrected sequences.
Ratatosk uses short and long reads to color paths in a compacted de Bruijn graph index and annotate vertices with candidate Single Nucleotide Polymorphisms.

□ malacoda: Bayesian modelling of high-throughput sequencing assays
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007504
malacoda, a statistical framework for the analysis of massively parallel genomic experiments which is designed to incorporate prior information in an unbiased way.
malacoda uses the negative binomial distribution with gamma priors to model sequencing counts while accounting for effects from input library preparation and sequencing depth.

□ The Cumulative Indel Model: fast and accurate statistical evolutionary alignment
>> https://academic.oup.com/sysbio/article/doi/10.1093/sysbio/syaa050/5870444
the probabilities of all possible alignments of all possible sequences will not sum up to one, but the probabilities of all alignments of the same length will.
The “cumulative indel model” approximates realistic evolutionary indel dynamics using differential equations. “Adaptive banding” reduces the computational demand of most alignment algorithms without requiring prior knowledge of divergence levels or pseudo-optimal alignments.

□ Discovering a sparse set of pairwise discriminating features in high dimensional data
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btaa690/5878953
define a class of problems in which linear separability of clusters is hidden in a low dimensional space. an unsupervised method to identify the subset of features that define a low dimensional subspace in which clustering can be conducted.
when the linear separability of clusters is restricted to a subspace, the identity of the subspace can be found without knowing the correct clusters by averaging over discriminators trained on an ensemble of proposed clustering configurations.
Crucially, eliminating any informative dimensions decreases the D/Ds ratio, moving to a regime in which conventional methods are more effective. it’s possible to artificially increase data density, and mitigate associated problems that are prevalent in high dimensional inference.

□ FIST: Imputation of Spatially-resolved Transcriptomes by Graph-regularized Tensor Completion
>> https://www.biorxiv.org/content/10.1101/2020.08.05.237560v1.full.pdf
The comprehensive evaluation of FIST on ten 10x Genomics Visium spatial genomics datasets and comparison with the methods for single-cell RNA sequencing data imputation demonstrate that FIST is a better method more suitable for spatial gene expression imputation.
FIST models sptRNA-seq data as a 3-way sparse tensor in genes and the spatial coordinates of the observed gene expressions, and then consider the imputation of the unobserved entries as a tensor completion problem in Canonical Polyadic Decomposition form.
□ NanoReviser: An Error-correction Tool for Nanopore Sequencing Based on a Deep Learning Algorithm
>> https://www.biorxiv.org/content/10.1101/2020.07.25.220855v1.full.pdf
NanoReviser, an open-source DNA basecalling reviser based on a deep learning algorithm to correct the basecalling errors introduced by current basecallers provided by default.
NanoReviser uses a CNN to extract the local patterns of the raw signals, and a highly powerful RNN and Bi-LSTMs to determine the long-term dependence of the bidirectional variation of the raw signals on DNA strand passing through the nanopore hidden in the basecalled sequences.
NanoReviser uses the Adam (adaptive moment estimation) algorithm with the default parameters in the training process to perform optimization. NanoReviser re-segmentes the raw electrical signals based on the basecalled sequences provided by the default basecallers.
□ DeepSF: A Deep Learning Framework for Predicting Human Essential Genes by Integrating Sequence and Functional data
>> https://www.biorxiv.org/content/10.1101/2020.08.04.236646v1.full.pdf
DeepSF can accurately predict human gene essentiality with an average performance of AUC about 94.35%, the area under precision-recall curve (auPRC) about 91.28%, the accuracy about 91.35%, and the F1 measure about 77.79%.
DeepSF is based on the multilayer perceptron structure. It uses ReLU as the activation function for all the hidden layers, while the output layer uses sigmoid activation function to perform discrete classification. The loss function in DeepSF is binary cross-entropy.

□ scRFE: Single-cell identity definition using random forests and recursive feature elimination
>> https://www.biorxiv.org/content/10.1101/2020.08.03.233650v1.full.pdf
scRFE (single-cell identity definition using random forests and recursive feature elimination) utilizes a random forest with recursive feature elimination and cross validation to identify each feature’s importance for classifying the input observations.
In order to learn the features to discriminate a given cell type from the others in the dataset, scRFE was built as a one versus all classifier. Recursive feature elimination was used to avoid high bias in the learned forest and to address multicollinearity.

□ Data-driven causal analysis of observational time series: a synthesis
>> https://www.biorxiv.org/content/10.1101/2020.08.03.233692v1.full.pdf
a synthesis of causal inference approaches including pairwise correlation and Reichenbach’s common cause principle, Granger causality, and state space reconstruction.
The problem of nonreverting continuous dynamics in state space reconstruction is similar to the non-stationarity problem in Granger causality, although they are distinct.
□ RSGSA: a Robust and Stable Gene Selection Algorithm
>> https://www.biorxiv.org/content/10.1101/2020.07.27.216879v1.full.pdf
Robust and stable gene selection algorithm (RSGSA) based on graph theory and ensembles of linear SVMs. At the beginning, highly correlated genes are discarded by employing a novel graph theoretic algorithm.
Stability of SVM-RFE is ensured by small noise in phenotypes. Symmetric uncertainty, gain ratio, Kullback-Leibler divergence, and RELIEF were used to evaluate the performance of RSGSA. Robustness is secured by instance level perturbation i.e, bootstrapping samples multiple times.
□ Dynamic regulatory module networks for inference of cell type specific transcriptional networks
>> https://www.biorxiv.org/content/10.1101/2020.07.18.210328v1.full.pdf
Dynamic Regulatory Module Networks (DRMNs) learn a cell type’s regulatory network from input expression and epigenomic profiles using multi-task learning to exploit cell type relatedness.
DRMNs are based on a non-stationary probabilistic model and can be used to model GRN on a lineage. DRMN inference runs for a set number of iterations or until convergence. Final module assignments are computed as maximum likelihood assignments using a dynamic programming.
□ METAWORKS: A flexible, scalable bioinformatic pipeline for multi-marker biodiversity assessments
>> https://www.biorxiv.org/content/10.1101/2020.07.14.202960v1.full.pdf
MetaWorks consists of a Conda environment and Snakemake pipeline that is meant to be run at the command line to bioinformatically processes Illumina paired-end metabarcodes from raw reads through to taxonomic assignments.
MetaWorks will fill a need in multi-marker metabarcoding studies that target taxa from multiple different domains of life, to provide a unified processing environment, pipeline, and taxonomic assignment approach for each marker from ribosomal RNA genes, spacers, or protein coding genes.
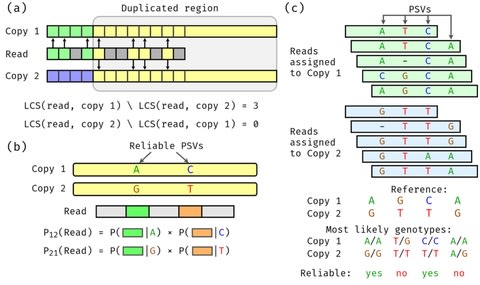
□ Sensitive alignment using paralogous sequence variants improves long read mapping and variant calling in segmental duplications
>> https://www.biorxiv.org/content/10.1101/2020.07.15.202929v1.full.pdf
DuploMap analyzes reads mapped to segmental duplications using existing long-read aligners and leverages paralogous sequence variants (PSVs) – sequence differences between paralogous sequences – to distinguish between multiple alignment locations.
DuploMap jointly performs read mapping and PSV genotyping using an iterative algorithm. DuploMap first retrieves all reads for which the primary alignment intersect the genomic intervals contained in the cluster. Next, it performs the following steps on the set of reads.

□ partR2: Partitioning R2 in generalized linear mixed models
>> https://www.biorxiv.org/content/10.1101/2020.07.26.221168v1.full.pdf
partR2 also estimates structure coefficients as the correlation between a predictor and fitted values, which provide an estimate of the total contribution of a fixed effect to the overall prediction, independent of other predictors.
partR2 implements parametric bootstrapping to quantify confidence intervals for each estimate. with real example datasets for Gaussian and binomials GLMMs and discuss interactions, which pose a specific challenge for partitioning the explained variance among predictors.

□ Noise regularization removes correlation artifacts in single-cell RNA-seq data preprocessing
>> https://www.biorxiv.org/content/10.1101/2020.07.29.227546v1.full.pdf
scRNA-seq data is further complicated by high dropout rate, which refers to the phenomenon by which a large proportion of genes have a measured read count of zero due to technical limitation in detecting the transcripts rather than true absence of the gene.
a model-agnostic noise regularization method that can effectively eliminate the correlation artifacts. False correlations from the overly smoothed data can be eliminated by the added noise while the true correlations should be robust enough to tolerate.
□ netAE: Semi-supervised dimensionality reduction of single-cell RNA sequencing to facilitate cell labeling
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btaa669/5877940
a network-enhanced autoencoder (netAE) aims to facilitate cell labeling with a semi-supervised method in an alternative pipeline, in which a few gold-standard labels are first identified and then extended to the rest of the cells computationally.
netAE outperforms various dimensionality reduction baselines and achieves satisfactory classification accuracy even when the labeled set is very small, without disrupting the similarity structure of the original space.
□ Uncovering Transcriptional Dark Matter via Gene Annotation Independent Single-Cell RNA Sequencing Analysis
>> https://www.biorxiv.org/content/10.1101/2020.07.31.229575v1.full.pdf
TAR-scRNA-seq (Transcriptionally Active Region single-cell RNA-seq) is a workflow that enables the discovery of transcripts beyond those listed in gene annotations.
TARs identified using the groHMM algorithm were labelled as annotated TAR or unannotated TAR features based on their overlap with existing gene annotations. These labeled TARs are then used to generate a TAR feature expression matrix in parallel with a gene expression matrix.
□ CellPhy: accurate and fast probabilistic inference of single-cell phylogenies from scDNA-seq data
>> https://www.biorxiv.org/content/10.1101/2020.07.31.230292v1.full.pdf
CellPhy is using bundled RAxML-NG, and capitalizes on numerous optimizations incl highly efficient and vectorized likelihood calculation code, coarse- and fine-grained parallelization with multi-threading and fast transfer bootstrap computation.
CellPhy evolves single-cell diploid DNA genotypes along the simulated genealogies under different scenarios including infinite- and finite-sites nucleotide mutation models, trinucleotide mutational signatures, sequencing and amplification errors.
CellPhy is based on a finite-site Markov nucleotide substitution model with 10 diploid states, and adopts the genotype equivalent of the classical general time-reversible (GTR) model of nucleotide substitution.

□ Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02100-5
knowledge-primed neural networks (KPNNs) exploits the ability of deep learning algorithms to assign meaningful weights in multi-layered networks, resulting in a widely applicable approach for interpretable deep learning.
KPNNs defines the cell surface receptor(s) expected to be most relevant for the biological phenomenon of interest, and extract a directed acyclic graph that connects the selected receptor(s) to all reachable transcription factors.
□ Probability-based methods for outlier detection in replicated high-throughput biological data
>> https://www.biorxiv.org/content/10.1101/2020.08.07.240473v1.full.pdf
an approach that accounts for technical variability and potential asymmetry that arise naturally in the distribution of replicate data, and aids in the identification of outliers.
Ideally, one would use exponential model-based methods when there are enough data points where computational intensity becomes an issue but when one still wants more accuracy than the original asymmetric Laplace-Weibull method.
□ IOEM: Efficient inference in state-space models through adaptive learning in online Monte Carlo expectation maximization
>> https://link.springer.com/article/10.1007/s00180-019-00937-4
IOEM can be applied with minimal prior knowledge of the model’s behavior, and requires no user supervision, while retaining the convergence guarantees of BEM/OEM, therefore providing an efficient, practical approach to parameter estimation in SMC methods.
a 2-dimensional autoregressive model and the stochastic volatility model to show the benefit of the proposed algorithm when inferring many parameters. IOEM produces accurate and precise parameter estimates when applied to continuous state-space models.
□ MultiPaths: a Python framework for analyzing multi-layer biological networks using diffusion algorithms
>> https://www.biorxiv.org/content/10.1101/2020.08.12.243766v1.full.pdf
Numerous methods for network analysis derived from graph theory have been adapted for a broad range of applications in the biomedical domain including target prioritization, gene prediction and patient stratification.
MultiPaths conducts several diffusion experiments on three independent multi-omics datasets over disparate networks generated from pathway databases, thus, highlighting the ability of multi-layer networks to integrate multiple modalities.

□ circHiC: circular visualization of Hi-C data and integration of genomic data
>> https://www.biorxiv.org/content/10.1101/2020.08.13.249110v1.full.pdf
The possibility to overlay genomic information aims at facilitating the exploration and understanding of chromosome structuring data.
The symmetry/redundancy property is conserved by default in circhic. Just as with the square matrix, this is particularly useful to highlight chromosome interaction domains. a “circle” corresponds to all contacts between pairs of loci separated by the same genomic distance.
□ glmGamPoi: Fitting Gamma-Poisson Generalized Linear Models on Single Cell Count Data
>> https://www.biorxiv.org/content/10.1101/2020.08.13.249623v1.full.pdf
Existing implementations for inferring its parameters from data often struggle with the size of single cell datasets, which typically comprise thousands or millions of cells; they do not take full advantage of the fact that zero and other small numbers are frequent in the data.
glmGamPoi provides inference of Gamma-Poisson generalized linear models with the following improvements over edgeR and DESeq2. glmGamPoi also provides a quasi-likelihood ratio test with empirical Bayesian shrinkage to identify differentially expressed genes.
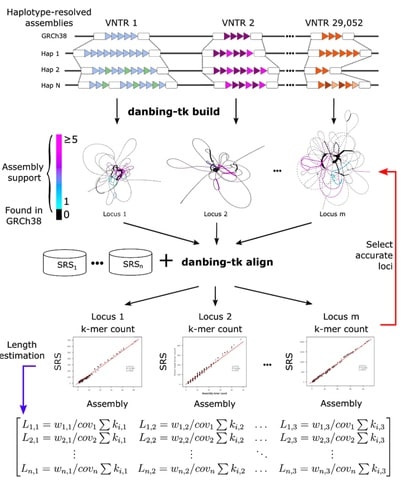
□ danbing-tk: Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs
>> https://www.biorxiv.org/content/10.1101/2020.08.13.249839v1.full.pdf
Solving the VNTR mapping problem for short reads by representing a collection of genomes with a repeat-pangenome graph, a data structure that encodes both the population diversity and repeat structure of VNTR loci.
Using long-read assemblies as ground truth, it is able to determine which VNTR loci may be accurately profiled using repeat-pangenome graph analysis with short reads.
Tandem Repeat Genotyping based on Haplotype-derived Pangenome Graphs (danbing-tk) to identify VNTR boundaries in assemblies, construct RPGGs, align SRS reads to the RPGG, and infer VNTR motif composition.

□ SQMtools: automated processing and visual analysis of ’omics data with R and anvi’o
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03703-2
SQMtools, a workflow that relies on the SqueezeMeta software for the automated processing of raw reads into annotated contigs and reconstructed genomes.
This engine allows users to input complex queries for selecting the contigs to be displayed based on their taxonomy, functional annotation and abundance across the different samples.

□ simATAC: A Single-cell ATAC-seq Simulation Framework
>> https://www.biorxiv.org/content/10.1101/2020.08.14.251488v1.full.pdf
simATAC simulates the library size using a bimodal GMM for samples from all platforms, and for non-10xG samples, the weight of the second Gaussian distribution can be set to zero.
simATAC estimates the proportion of cells with non-zero entries for the jth bin, pj, based on the user-input real scATAC-seq bin by cell matrix, and determines if an entry’s status in the simulated count matrix is zero or non-zero based on a Bernoulli distribution.

□ ACValidator: A novel assembly-based approach for in silico verification of circular RNAs
>> https://academic.oup.com/biomethods/article/5/1/bpaa010/5849853
ACValidator takes as input a sequence alignment mapping (SAM) file and the circRNA coordinate(s) to be validated.
ACValidator operates in three phases: (i) extraction and assembly of reads from the SAM file to generate contigs; (ii) generation of a pseudo-reference file; and (iii) alignment of contigs from Phase 1 against the pseudo-reference from Phase 2.
□ CIPHER-SC: Disease-Gene Association Inference Using Graph Convolution on a Context-Aware Network with Single-Cell Data
>> https://ieeexplore.ieee.org/document/9170857
CIPHER-SC, a graph convolution-based approach to realize a complete end-to-end learning architecture. CIPHER-SC constructs a context-aware network to unbiasedly integrate all data sources.
CIPHER-SC shows that its complete end-to-end design and unbiased data integration boost the performance from 0.8727 to 0.9443 in AUC.
 (photo by
(photo by