Der Mond war immer und immer wieder, aber der Mond gibt es nicht mehr zurück.

□ Genotype–phenotype mapping in another dimension:
>> https://www.nature.com/articles/s41576-019-0170-y
Exploring genetic interaction manifolds constructed from rich single-cell phenotypes.
an analytical framework for interpreting high-dimensional landscapes of cell states (manifolds) constructed from transcriptional phenotypes.
□ NExUS: Bayesian simultaneous network estimation across unequal sample sizes
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz636/5555873
when estimating multiple networks across heterogeneous sub-populations, varying sample sizes pose a challenge in the estimation and inference, as network differences may be driven by differences in power.
NExUS (Network Estimation across Unequal Sample sizes), a Bayesian method that enables joint learning of multiple networks while avoiding artefactual relationship between sample size and network sparsity.

□ LUCID: A Latent Unknown Clustering Integrating Multi-Omics Data with Phenotypic Traits
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz667/5556107
Latent Unknown Clustering with Integrated Data (LUCID), to perform integrative analyses using multi-om- ics data with phenotypic traits, leveraging their underlying causal relationships in latent cluster estimations.
The LUCID framework allows for flexibility by permitting every conditional distribution to vary according to specific characteristics defined in the linear predictors or in the link functions to the outcome.

□ ScisTree: Accurate and Efficient Cell Lineage Tree Inference from Noisy Single Cell Data: the Maximum Likelihood Perfect Phylogeny Approach
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz676/5555811
ScisTree ({S}ingle {c}ell {i}nfinite {s}ites {Tree}) assumes the infinite sites model, and inferring cell tree and calling genotypes from uncertain single cell genotype data.
ScisTree implements a fast heuristic for finding the cell lineage tree that maximizes the genotype probability under the infinite sites model over the tree space.


□ Sampling rare events across dynamical phase transitions
>> https://aip.scitation.org/doi/full/10.1063/1.5091669
the application of a particular rare-event simulation technique, based on cloning Monte Carlo methods, to characterize dynamical phase transitions in paradigmatic stochastic lattice gases.
These models exhibit spontaneous breaking of time-translation symmetry at the trajectory level under periodic boundary conditions, via the appearance of a time-dependent traveling wave.
□ Hilbert spaces and C*-algebras are not finitely concrete
>> https://arxiv.org/pdf/1908.10200v1.pdf
Finite concreteness of a category is an essential first test in determining the extent to which it can be subjected to a model-theoretic analysis.
any category axiomatizable in the infinitary logic L∞,ω (i.e. (∞,ω)-elementary is finitely concrete.
that not only is Hilbr not an AEC with respect to the usual forgetful functor—which is obvious from the failure of the union of chains axiom—this problem is essential: Hilbr is not equivalent to an AEC, or an elementary category.
□ RNNs Evolving on an Equilibrium Manifold: A Panacea for Vanishing and Exploding Gradients?
>> https://arxiv.org/pdf/1908.08574.pdf
Equilibriated Recurrent Neural Networks (ERNNs) that overcome the gradient decay or explosion effect and lead to recurrent models that evolve on the equilibrium manifold.
ERNNs account for long-term dependencies, and can efficiently recall informative aspects of data from the distant past.
A key insight is to respond to a new signal input, by updating the state-vector that corresponds to a point on the manifold, rather than taking a direction pointed to by the vector-field.

□ A new variance ratio metric to detect the timescale of compensatory dynamics
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/28/742510.full.pdf
a new timescale-specific variance ratio appropriate for terrestrial grasslands and other systems with shorter, regularly-spaced time series.
addressing the fundamental questions of whether synchrony/compensatory dynamics are timescale-dependent phenomena, and whether compensatory dynamics are rare, compared to synchrony.

□ Epi-GTBN: an approach of epistasis mining based on genetic Tabu algorithm and Bayesian network
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3022-z
In order to solve the search stagnant and premature phenomenon that generated by general crossover operators, Epi-GTBN uses the memory function of tabu search method into the crossover operation of genetic algorithm.
Epi-GTBN converts the genotypic data into binary Boolean data, and carries out the fast logic (bitwise) operation directly to calculate the mutual information.

□ A new grid- and modularity-based layout algorithm for complex biological networks
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0221620
GML is a novel layout algorithm that aims to clarify the network complexity according to its inherent modular structure.
With the VisANT plugin of GML, researchers can gain insights into global characteristics as well as discern network details of biological networks.

□ GeneWalk identifies relevant gene functions for a biological context using network representation learning https://www.biorxiv.org/content/biorxiv/early/2019/09/05/755579.full.pdf
Networks of biological mechanisms are now available from knowledge bases such as Pathway Commons, String, Omnipath, and the Integrated Network and Dynamical Reasoning Assembler (INDRA).
After automatic assembly of an experiment-specific gene regulatory network, GeneWalk quantifies the similarity between vector representations of each gene and its GO annotations through representation learning.

□ Genomic GPS: using genetic distance from individuals to public data for genomic analysis without disclosing personal genomes
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1792-2
Genomic global positioning system (GPS) applies the multilateration technique commonly used in the GPS to genomic data.
derived a mathematical proof showing that in N-dimensional space, and with K reference nodes with known positions, an unknown node’s coordinates can be unequivocally identified if K > N.
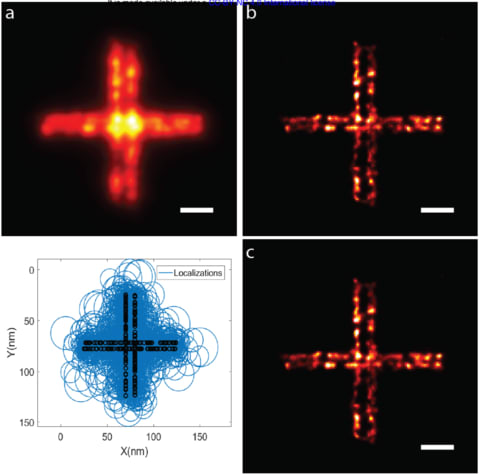
□ Sub-Nanometer Precision using Bayesian Grouping of Localizations
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/30/752287.full.pdf
a Bayesian method of grouping and combining localizations from multiple blinking/binding events that can improve localization precision to better than one nanometer.
The known statistical distribution of the number of binding/blinking events per dye/docking strand along with the precision of each localization event are used to estimate the true number and location of emitters in closely-spaced clusters.
□ Calculating power for the general linear multivariate model with one or more Gaussian covariates
>> https://www.tandfonline.com/doi/full/10.1080/03610926.2018.1433849
The new method approximates the noncentrality parameter under the alternative hypothesis using a Taylor series expansion for the matrix-variate beta distribution of type I.
a noncentral F power approximation for hypotheses about fixed predictors in general linear multivariate models with one or more Gaussian covariates.
□ qtQDA: quantile transformed quadratic discriminant analysis for high-dimensional RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/31/751370.full.pdf
a new classification method based on a model where the data is marginally negative binomial but dependent, thereby incorporating the dependence known to be present between measurements from different genes.
qtQDA works by first performing a quantile transformation (qt) then applying Gaussian Quadratic Discriminant Analysis (QDA) using regularized covariance matrix estimates.

□ Robustness and applicability of functional genomics tools on scRNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/01/753319.full.pdf
The bulk tools are PROGENy, DoRothEA and classical GO enrichment analysis combining GO gene sets with GSEA. PROGENy estimates the activity of 14 signaling pathways by combining corresponding gene sets with a linear model.
the D-AUCell and metaVIPER performed better on single cells than on the original bulk samples. metaVIPER used the same statistical method as DoRothEA but different gene set resources.

□ MPRAnalyze: statistical framework for massively parallel reporter assays:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1787-z
MPRAnalyze leverages the unique structure of MPRA data to quantify the function of regulatory sequences, compare sequences’ activity across different conditions, and provide necessary flexibility in an evolving field.
This framework comprises 2 nested models: the DNA model, which estimates the latent construct counts for the observed DNA counts, and the RNA model, which uses the construct count estimates from the DNA model and the observed RNA counts to estimates the rate of transcription, α.

□ Cytoscape Automation: empowering workflow-based network analysis
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1758-4
Cytoscape Automation (CA), which marries Cytoscape to highly productive workflow systems, for example, Python/R in Jupyter/RStudio.
a bioinformatician can create novel network biologic workflows as orchestrations of Cytoscape functions, complex custom analyses, and best-of-breed external tools and language-specific libraries.
enable Commands and Cytoscape apps to be called through CyREST and encourage high-quality documentation of CyREST endpoints using state-of-the-art documentation systems (such as Swagger) and interactive call prototyping.

□ CNEr: A toolkit for exploring extreme noncoding conservation
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006940
Clusters of CNEs coincide with topologically associating domains (TADs), indicating ancient origins and stability of TAD locations.
This has suggested further hypotheses about the still elusive origin of CNEs, and has provided a comparative genomics-based method of estimating the position of TADs around developmentally regulated genes in genomes where chromatin conformation capture data is missing.
□ BONITA: Executable pathway analysis using ensemble discrete-state modeling for large-scale data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007317
Boolean Omics Network Invariant-Time Analysis (BONITA), for signal propagation, signal integration, and pathway analysis.
BONITA’s signal propagation approach models heterogeneity in transcriptomic data as arising from intercellular heterogeneity rather than intracellular stochasticity, and propagates binary signals repeatedly across networks.

□ NPA: an R package for computing network perturbation amplitudes using gene expression data and two-layer networks https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3016-x
The NPA (Network Perturbation Amplitude) and BIF (Biological Impact Factor) methods allow to understand the mechanisms behind and predict the effect of exposure based on transcriptomics datasets.
The functionalities are implemented using R6 classes, making the use of the package seamless and intuitive. The various network responses are analyzed using the leading node analysis, and an overall perturbation, called the Biological Impact Factor, is computed.
The NPA package implements the published network perturbation amplitude methodology and provides a set of two-layer networks encoded in the Biological Expression Language.
□ DENOPTIM: Software for Computational de Novo Design of Organic and Inorganic Molecules
>> https://pubs.acs.org/doi/10.1021/acs.jcim.9b00516
DENOPTIM (De Novo OPTimization of In/organic Molecules) is a software meant for de novo design and virtual screening of functional compounds.
In practice, DENOPTIM is meant for building chemical entities by assembling building blocks (i.e., fragments), processing each chemical entity as to produce its figure of merit (i.e., fitness), and designing new entities based on the properties of entities generated before.

□ rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data
>> https://academic.oup.com/gigascience/article/8/9/giz100/5559527
Although every transcriptome assembler presented in this study has its own benefits and drawbacks, the trade-off between assembly completeness and correctness can be significantly shifted by modifying the algorithms’ parameters.
the novel transcriptome assembler rnaSPAdes, which has been developed on top of the SPAdes genome assembler and explores computational parallels between assembly of transcriptomes and single-cell genomes.

□ EnClaSC: A novel ensemble approach for accurate and robust cell-type classification of single-cell transcriptomes
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/04/754085.full.pdf
EnClaSC draws on the idea of ensemble learning in the feature selection, few-sample learning, neural network and joint prediction modules, respectively, and thus constitutes a novel ensemble approach for cell-type classification of single-cell transcriptomes.
EnClaSC can not only be applied to the self-projection within a specific dataset and the cell-type classification across different datasets, but also scale up well to various data dimensionality and different data sparsity.
□ MLDSP-GUI: An alignment-free standalone tool with an interactive graphical user interface for DNA sequence comparison and analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/745406.full.pdf
MLDSP-GUI (Machine Learning with Digital Signal Processing) is an open-source, alignment- free, ultrafast, computationally lightweight, standalone software tool with an interactive Graphical User Interface for comparison and analysis of DNA sequences.
MLDSP-GUI combines both approaches in that it can use one-dimensional numerical representations of DNA sequences that do not require calculating k-mer frequencies, but, in addition, it can also use k-mer dependent two-dimensional Chaos Game Representation of DNA sequences.

□ NIHBA: A Network Interdiction Approach with Hybrid Benders Algorithm for Strain Design
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/04/752923.full.pdf
a network interdiction model free of growth optimality assumptions, a special case of bilevel optimisation, for computational strain design and have developed a hybrid Benders algorithm (HBA).
that deals with complicating binary variables in the model, thereby achieving high efficiency without numeric issues in search of best design strategies.

□ ADAPT-CAGE: Solving the transcription start site identification problem with a Machine Learning algorithm for analysis of CAGE data
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/04/752253.full.pdf
ADAPT-CAGE, a Machine Learning framework which is trained to distinguish between CAGE signal derived from TSSs and transcriptional noise. ADAPT-CAGE provides annotation-agnostic, highly accurate and single-nucleotide resolution experimentally derived TSSs on a genome-wide scale.
ADAPT-CAGE exhibits improved performance on every benchmark that we designed based on both annotation- and experimentally-driven strategies.
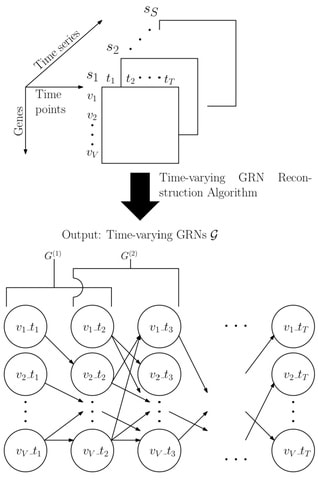
□ Rapid Reconstruction of Time-varying Gene Regulatory Networks with Limited Main Memory
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/755249.full.pdf
TGS-Lite provides the same time complexity and true positive detection power as those of TGS at a significantly lower memory requirement, that grows linearly to the number of genes.
Similarly, TGS- Lite+ offers the superior time complexity and reconstruction power of TGS+ with a linear memory requirement.
the time-varying GRN structures are reconstructed independently of each other. Thus, the framework is compatible with any time-series gene expression dataset, regardless of whether the true GRNs follow the smoothly time- varying assumption or not.

□ A framework for quantifying deviations from dynamic equilibrium theory
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/755645.full.pdf
Community assembly is governed by colonization and extinction processes, and the simplest model describing it is Dynamic Equilibrium (DE) theory, which assumes that communities are shaped solely by stochastic colonization and extinction events.
The PARIS model satisfies the assumptions of Dynamic Equilibrium and can be used to generate multiple synthetic time series that 'force' these assumptions on the data.
□ Toward a dynamic threshold for quality-score distortion in reference-based alignment
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/754614.full.pdf
While the ultimate interest lies in assessing the impact of lossy quality score representation in a full bioinformatic pipeline, this approach may be ineffective for the purpose of understanding the role of lossy quality scores in the analysis.
This is because sequence data is transformed continually as it is shepherded throughout the pipeline, and errors and associated uncertainties that operate on it are combined along with the data, obscuring the precise effect of lossy quality scores in the analysis.
□ Omic-Sig: Utilizing Omics Data to Explore and Visualize Kinase-Substrate Interactions
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/746123.full.pdf
the information gained via interrogation of global proteomes and transcriptomes can offer additional insight into the interaction of kinases and their respective substrates.
Omic-Sig is a bioinformatics tool to stratify phospho-substrates and their associated kinases by utilizing the differential abundances between case and control samples in phosphoproteomics, global proteomics, and transcriptomics data.

□ Whole Genome Tree of Life: Deep Burst of Organism Diversity
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/756155.full.pdf
An organism Tree of Life (organism ToL) is a conceptual and metaphorical tree to capture a simplified narrative of the evolutionary course and kinship among the extant organisms of today.
whole genome information can be compared without sequence alignment, all extant organisms can be classified into six large groups and all the founders of the groups have emerged in a Deep Burst at the very beginning period of the emergence of the Life on Earth.

□ Contour Monte Carlo: A Monte Carlo method to estimate cell population heterogeneity
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/758284.full.pdf
a computational sampling method named “Contour Monte Carlo” for estimating mathematical model parameters from snapshot distributions, which is straightforward to implement and does not require cells be assigned to predefined categories.
Contour Monte Carlo provides an automatic framework for performing inference on such under-determined systems, and the use of priors allows for robust and precise parameter estimation unattainable through the data alone.

□ GPU Accelerated Adaptive Banded Event Alignment for Rapid Comparative Nanopore Signal Analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/756122.full.pdf
comparing raw nanopore signals to a biological reference sequence is a computationally complex task despite leveraging a dynamic programming algorithm for Adaptive Banded Event Alignment (ABEA)—a commonly used approach to polish sequencing data and identify non-standard nucleotides.
The re-engineered version of the Nanopolish methylation detection module, f5c that employs the GPU accelerated Adaptive Banded Event Alignment was not only around 9× faster on an HPC, but also reduced the peak RAM by around 6× times.

□ Non-homogeneous dynamic Bayesian networks with edge-wise sequentially coupled parameters
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz690/5561098
As the enforced similarity of the network parameters can have counter-productive effects, a new consensus NH-DBN model that combines features of the uncoupled and the coupled NH-DBN.
The new model infers for each individual edge whether its interaction parameter stays similar over time (and should be coupled) or if it changes from segment to segment.
□ CroP - Coordinated Panel Visualization for Biological Networks Analysis
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz688/5559487
CroP is a data visualization application that focuses on the analysis of relational data that changes over time.
While it was specifically designed for addressing the preeminent need to interpret large scale time series from gene expression studies, CroP is prepared to analyze datasets from multiple contexts.
Through clustering and the time curve visualization it is possible to quickly identify groups of data points with similar proprieties or behaviours, as well as temporal patterns across all points, such as periodic waves of expression.

□ Splotch: Robust estimation of aligned spatial temporal gene expression data
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/757096.full.pdf
describing computational methods matched to ST technology for interrogating spatiotemporal dynamics of diseases, cell-cell communication, and regulatory dynamics in complex tissues.
Splotch, a novel computational framework for the analysis of spatially resolved transcriptomics data. Splotch aligns transcriptomics data from multiple tissue sections and timepoints to generate improved posterior estimates of gene expression.


□ NLIMED: Natural Language Interface for Model Entity Discovery in Biosimulation Model Repositories
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/05/756304.full.pdf
Natural Language Interface for Model Entity Discovery (NLIMED) is an interface to search model entity (i.e. flux of sodium across basolateral plasma membrane, concentration of potassium in the portion of tissue fluid) from the collection of biosimulation models in repository.
NLIMED works by converting natural language query into SPARQL, so it may help researcher to avoid the rigid syntax of SPARQL, query path consisting multiple predicates, and detail description of class ontologies.
□ sketchy
>> https://github.com/esteinig/sketchy
Real-time lineage hashing and genotyping of bacterial pathogens from uncorrected nanopore reads