









□ End-to-end Learning of Evolutionary Models to Find Coding Regions in Genome Alignments
>> https://www.biorxiv.org/content/10.1101/2021.03.09.434414v1.full.pdf
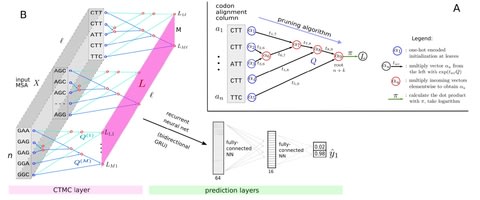
ClaMSA (Classify Multiple Sequence Alignments) uses the standard general-time reversible (GTR) CTMC on a tree. ClaMSA outperforms both the dN/dS test and PhyloCSF by a wide margin in the task of codon alignment classification.
Even of higher meaning could be the general-time reversible CTMC layer that allows to compute gradients of the tree-likelihood under the almost universally used continuous-time Markov chain model.
□ Cobolt: Joint analysis of multimodal single-cell sequencing data
>> https://www.biorxiv.org/content/10.1101/2021.04.03.438329v1.full.pdf
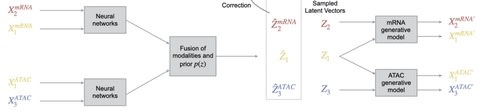
Cobolt integrates multi-modality platforms with single-modality platforms by jointly analyzing a SNARE-seq dataset, a single-cell gene expression dataset, and a single-cell chromatin accessibility dataset.
Cobolt’s generative model for a single modality i starts by assuming that the counts measured on a cell are the mixture of the counts from different latent categories. Cobolt results in an estimate of the latent variable zc for each cell, which is a vector that lies in a K-dimensional space.
□ superSTR: Ultrafast, alignment-free detection of repeat expansions in NGS and RNAseq data
>> https://www.biorxiv.org/content/10.1101/2021.04.05.438449v1.full.pdf
superSTR uses a fast, compression-based estimator of the information complexity of individual reads to select and process only those reads likely to harbour expansions.
superSTR identifies samples with REs and to screen motifs for expansion in raw sequencing data from short-read WGS experiments, in biobank-scale analysis, and for the first time in direct interrogation of repeat sequences.
□ OBSDA: Optimal Bayesian supervised domain adaptation for RNA sequencing data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab228/6211157
OBSDA provides an efficient Gibbs sampler for parameter inference. And leverages the gene-gene network prior information. OBSDA can be applied in cases where different domains share the same labels or have different ones.
OBSDA is based on a hierarchical Bayesian negative binomial model with parameter factorization, for which the optimal predictor can be derived by marginalization of likelihood over the posterior of the parameters.
□ Ordmeta: Powerful p-value combination methods to detect incomplete association
>> https://www.nature.com/articles/s41598-021-86465-y
Weighted Fisher’s method (wFisher) uses a gamma distribution to assign non-integer weights to each p-value that are proportional to sample sizes, while the total weight is kept as small as that of Fisher’s method (2n).
Ordmeta calculates p-value for the minimum marginal p-value. In other words, it assesses the positions of each marginal statistic p(i) to select the optimal one and assess its significance using joint distribution of order statistic.

□ Hubness reduction improves clustering and trajectory inference in single-cell transcriptomic data
>> https://www.biorxiv.org/content/10.1101/2021.03.18.435808v1.full.pdf
Investigate the phenomenon of hubness in scRNA-seq data in spaces of increasing dimensionality. Certain manifestations of the dimensionality curse might appear starting with an intrinsic dimensionality as low as 10.
By the reverse-coverage approach, Hubness reduction can be used instead of dimensionality reduction, in order to compensate for certain manifestations of the dimensionality curse using k-NN graphs or distance matrices as an essential ingredient.
□ Randomness extraction in computability theory
>> https://arxiv.org/pdf/2103.03971.pdf
The analysis of the extraction rates of these three classes of examples draws upon the machinery of effective ergodic theory, using certain effective versions of Birkhoff’s ergodic theorem.
For the limn→∞ Avg(φ, μ, n) to exist, the function φ must be regular in the relative amount of input needed for a given amount of output.
First, there are the so-called online continuous functions, which compute exactly one bit of output for each bit of input. On the other hand, there are the random continuous functions which produce regularity in a probabilistic sense.

□ RPVG: Haplotype-aware pantranscriptome analyses using spliced pangenome graphs
>> https://www.biorxiv.org/content/10.1101/2021.03.26.437240v1.full.pdf
VG RNA uses the Graph Burrows-Wheeler Transform (GBWT) to efficiently store the HST paths allowing the pipeline to scale to a pantranscriptome with millions of transcript paths.
VG MPMAP produces multipath alignments that capture the local uncertainty of an alignment to different paths in the graph. Lastly, the expression of the HSTs are inferred from the multipath alignments using RPVG.
RPVG uses a nested inference scheme that first samples the most probable underlying haplotype combinations (e.g. diplotypes) and then infers the HST expression using expectation maximization conditioned on the sampled haplotypes.
□ ZEAL: Protein structure alignment based on shape similarity:
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab205/6194581
ZEAL (ZErnike-based protein shape ALignment), an interactive tool to superpose global and local protein structures based on their shape resemblance using 3D functions to represent the molecular surface.
ZEAL uses Zernike-Canterakis functions to describe the shape of the molecular surface and provides an optimal superposition between two proteins by maximizing the correlation between the moments computed from these functions.

□ RENANO: a REference-based compressor for NANOpore FASTQ files
>> https://www.biorxiv.org/content/10.1101/2021.03.26.437155v1.full.pdf
Good compression results are obtained by keeping the positions of the reference base call strings that are used by at least two atomic alignments, with no significant improvement for larger thresholds.
RENANO, a lossless NPS FASTQ data compressor that builds on its predecessor ENANO, introducing two novel reference-based compres- sion algorithms for base call strings that significantly improve the state of the art compression performance.
RENANOind directly benefiting from having multiple atomic alignments that use the same sections of the reference strings, which is less likely to happen in files with low coverage.

□ Clustering and Recognition of Spatiotemporal Features Through Interpretable Embedding of Sequence to Sequence Recurrent Neural Networks
>> https://www.frontiersin.org/articles/10.3389/frai.2020.00070/full
Embedding space projections of the decoder states of RNN Seq2Seq model trained on sequences prediction are organized in clusters capturing similarities and differences in the dynamics of these sequences.
The embedding can be mapped through Proper Orthogonal Decomposition of concatenated encoder and decoder internal states. The encoder trajectory initiated from various starting points connects them in the interpretable embedding space with the appropriate decoder trajectory.
□ Information theoretic perspective on genome clustering
>> https://www.sciencedirect.com/science/article/pii/S1319562X20307038
Shannon’s information theoretic perspective of communication helps one to understand the storage and processing of information in these one-dimensional sequences.
There is an inverse correlation of the markovian contribution to the relative information content or Shannon redundancy arising from di and tri nucleotide arrangements (RD2 + RD3) with | %AT-50 |.

□ c-CSN: Single-cell RNA Sequencing Data Analysis by Conditional Cell-specific Network
>> https://www.sciencedirect.com/science/article/pii/S1672022921000589
c-CSN method, which can construct the conditional cell-specific network (CCSN) for each cell. c-CSN method can measure the direct associations between genes by eliminating the indirect associations.
c-CSN can be used for cell clustering and dimension reduction on a network basis of single cells. Intuitively, each CCSN can be viewed as the transformation from less “reliable” gene expression to more “reliable” gene-gene associations in a cell.
the network flow entropy (NFE) integrates the scRNA-seq profile of a cell with its gene-gene association network, and the results show that NFE performs well in distinguishing various cells of differential potency.
□ GRAMMAR-Lambda: An Extreme Simplification for Genome-wide Mixed Model Association Analysis
>> https://www.biorxiv.org/content/10.1101/2021.03.10.434574v1.full.pdf
At a moderate or genomic heritability, polygenic effects can be estimated using a small number of randomly selected markers, which extremely simplify genome-wide association analysis w/ an approximate computational complexity to naïve method in large-scale complex population.
GRAMMAR-Lambda adjusts GRAMMAR using genomic control, extremely simplifying genome-wide mixed model analysis. For a complex population structure, a high false-negative error of GRAMMAR can be efficiently corrected by dividing genome-wide test statistics by genomic control.

□ DCI: Learning Causal Differences between Gene Regulatory Networks
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab167/6168117
Difference Causal Inference (DCI) algorithm infers changes (i.e., edges that appeared, disappeared or changed weight) between two causal graphs given gene expression data from the two conditions.
DCI algorithm is efficient in its use of samples and computation since it infers the differences between causal graphs directly without estimating each possibly large causal graph separately.
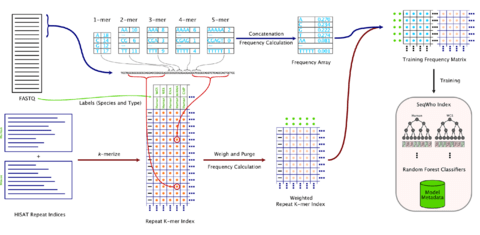
□ SeqWho: Reliable, rapid determination of sequence file identity using k-mer frequencies
>> https://www.biorxiv.org/content/10.1101/2021.03.10.434827v1.full.pdf
SeqWho is designed to heuristically assess the quality of sequencing and classify the organism and protocol type. This is done in an alignment-free algorithm that leverages a Random Forest classifier to learn from native biases in k-mer frequencies and repeat sequence identities.
□ TIGER: inferring DNA replication timing from whole-genome sequence data
>> https://pubmed.ncbi.nlm.nih.gov/33704387/
TIGER (Timing Inferred from Genome Replication), a computational approach for extracting DNA replication timing information from whole genome sequence data obtained from proliferating cell samples.
Replication dynamics can hence be observed in genome sequence data by analyzing DNA copy number along chromosomes while accounting for other sources of sequence coverage variation. TIGER is applicable to any species with a contiguous genome assembly and rivals the quality of experimental measurements of DNA replication timing.
□ CONSULT: Accurate contamination removal using locality-sensitive hashing
>> https://www.biorxiv.org/content/10.1101/2021.03.18.436035v1.full.pdf
CONSULT has higher true-positive and lower false-positive rates of contamination detection than leading methods such as Kraken-II and improves distance calculation from genome skims.
CONSULT saves reference k-mers in a LSH-based lookup table. CONSULT may enable by allowing distant matches is inclusion filtering: find reads that seem to belong to the group of interest if assembled genomes from that phylogenetic group are available.

□ VeloAE: Representation learning of RNA velocity reveals robust cell transitions
>> https://www.biorxiv.org/content/10.1101/2021.03.19.436127v1.full.pdf
VeloAE can both accurately identify stimulation dynamics in time-series designs and effectively capture the expected cellular differentiation in different biological systems.
Cross-Boundary Direction Correctness (CBDir) and In-Cluster Coherence (ICVCoh), for scoring the direction correctness and coherence of estimated velocities. These metrics can complement the usual vague evaluation with mainly visual plotting of velocity filed.

□ SLR-superscaffolder: a de novo scaffolding tool for synthetic long reads using a top-to-bottom scheme
>> https://pubmed.ncbi.nlm.nih.gov/33765921/
SLR-superscaffolder requires an SLR dataset plus a draft assembly as input. A draft assembly can be a set of contigs or scaffolds pre-assembled by various types of datasets.
SLR-superscaffolder calculates the correlation between contigs to construct a scaffold graph to reduce the graph complexities caused by repeats. The number of iterations were set to avoid a possible significant reduction of connectivity in the co-barcoding scaffold graph.

□ KiMONo: Versatile knowledge guided network inference method for prioritizing key regulatory factors in multi-omics data
>> https://www.nature.com/articles/s41598-021-85544-4
KiMONo leverages various prior information, reduces the high dimensional input space, and uses sparse group LASSO (SGL) penalization in the multivariate regression approach to model each gene's expression level.
Within SGL, the parameters α denotes the intergroup penalization while τ defines the group-wise penalization. KiMONo approximates an optimal parameter setting via using the Frobenius norm.

□ BugSeq: a highly accurate cloud platform for long-read metagenomic analyses
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04089-5
On the ZymoBIOMICS Even and Log communities, BugSeq (F1 = 0.95 at species level) offers better read classification than MetaMaps (F1 = 0.89–0.94) in a fraction of the time.
BugSeq was found to outperform MetaMaps, CDKAM and Centrifuge, sometimes by large margins (up to 21%), in terms of precision and recall. BugSeq is an order of magnitude faster than MetaMaps, which took over 5 days using 32 cores and their “miniSeq + H” database.

□ A new algorithm to train hidden Markov models for biological sequences with partial labels
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04080-0
A novel Baum–Welch based HMM training algorithm to leverage partial label information with techniques of model selection through partial labels.
The constrained Baum–Welch algorithm (cBW) is similar to the standard Baum–Welch algorithm except that the training sequences are partially labelled, which imposes the constraints on the possible hidden state paths in calculating the expectation.
□ BayesASE: Testcrosses are an efficient strategy for identifying cis regulatory variation: Bayesian analysis of allele specific expression
>> https://academic.oup.com/g3journal/advance-article/doi/10.1093/g3journal/jkab096/6192811
BayesASE is a complete bioinformatics pipeline that incorporates state-of-the-art error reduction techniques and a flexible Bayesian approach to estimating Allelic imbalance (AI) and formally comparing levels of AI between conditions.
BayesASE consists of four main modules: Genotype Specific References, Alignment and SAM Compare, Prior Calculation, and Bayesian Model. The Alignment and SAM Compare module quantifies alignment counts for each input file for each of the two genotype specific genomes.
□ L2,1-norm regularized multivariate regression model with applications to genomic prediction
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab212/6198100
a L2,1-norm regularized multivariate regression model and devise a fast and efficient iterative optimization algorithm, called L2,1-joint, applicable in multi-trait GS.
The capacity for variable selection allows us to define master regulators that can be used in a multi-trait GS setting to dissect the genetic architecture of the analyzed traits.
the effectiveness of the L2,1-norm as a tool for variable selection and master regulators identification in a penalized multivariate regression when the number of SNPs, as predictors, is much larger than the number of genotypes.
□ Boosting heritability: estimating the genetic component of phenotypic variation with multiple sample splitting
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04079-7
the linear model that relates a trait with a genotype matrix, then narrow-sense heritability is defined together with some discussion regarding the fixed-effect vs. random-effect approach for estimation.
a generic strategy for heritability inference, termed as “boosting heritability”, by combining the advantageous features of different recent methods to produce an estimate of the heritability with a high-dimensional linear model.
□ The CINECA project: Biomedical Named entity recognition - Pros and cons of rule-based and deep learning methods
>> https://www.cineca-project.eu/blog-all/biomedical-named-entity-recognition-pros-and-cons-of-rule-based-and-deep-learning-methods
To create a standardised metadata representation CINECA is using Natural language processing (NLP) techniques such as entity recognition, using rule-based tools such as MetaMap, LexMapr, and Zooma.

□ ModPhred: an integrative toolkit for the analysis and storage of nanopore sequencing DNA and RNA modification data
>> https://www.biorxiv.org/content/10.1101/2021.03.26.437220v1.full.pdf
ModPhred integrates probabilistic DNA and RNA modification information within the FASTQ and BAM file formats, can be used to encode multiple types of modifications simultaneously, and its output can be easily coupled to genomic track viewers.
ModPhred can extract and encode modification information from basecalled FAST5 datasets 4-8 times faster than Megalodon, while producing output files that are 50 times smaller.
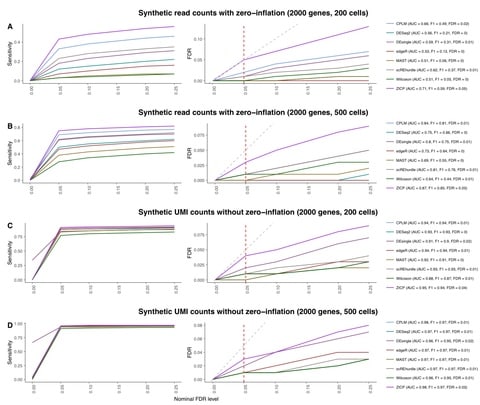
□ Differential expression of single-cell RNA-seq data using Tweedie models
>> https://www.biorxiv.org/content/10.1101/2021.03.28.437378v1.full.pdf
Tweedieverse can flexibly capture a large dynamic range of observed scRNA-seq data across experimental platforms induced by heavy tails, sparsity, or different count distributions to model the technological variability in scRNA-seq expression profiles.
the zero-inflated Tweedie model as Zero-inflated Compound Poisson Linear Model (ZICP) that allows zero probability mass to exceed a traditional Tweedie distribution to model zero-inflated scRNA-seq data with excessive zero counts.
□ EVI: Evidence Graphs: Supporting Transparent and FAIR Computation, with Defeasible Reasoning on Data, Methods and Results
>> https://www.biorxiv.org/content/10.1101/2021.03.29.437561v1.full.pdf
EVI integrates FAIR practices on data and software, with important concepts from provenance models, and argumentation theory. It extends PROV for additional expressiveness, with support for defeasible reasoning.
EVI is an extension of W3C PROV, based on argumentation theory. Evidence Graphs are directed acyclic graphs. They are first-class digital objects and may have their own persistent identifiers and be referenced as part of the metadata of any result.

□ CIDER: An interpretable meta-clustering framework for single-cell RNA-Seq data integration and evaluation
>> https://www.biorxiv.org/content/10.1101/2021.03.29.437525v1.full.pdf
The core of CIDER is the IDER metric, which can be used to compute the similarity between two groups of cells across datasets. Differential expression in IDER is computed using limma-voomor limma-trend which was chosen from a collection of approaches for DE analysis.
CIDER used a novel and intuitive strategy that measures the similarity by performing group- level calculations, which stabilize the gene-wise variability. CIDER can also be used as a ground-truth-free evaluation metric.
□ DISTEMA: distance map-based estimation of single protein model accuracy with attentive 2D convolutional neural network
>> https://www.biorxiv.org/content/10.1101/2021.03.29.437573v1.full.pdf
DISTEMA comprises multiple convolutional layers, batch normalization layers, dense layers, and Squeeze-and-Excitation blocks with attention to automatically extract features relevant to protein model quality from the raw input without using any expert-curated features.
DISTEMA performed better than QDeep according to the ranking loss even though it only used one kind of input information, but worse than QDeep according to Pearson’s correlation.

□ An introduction to new robust linear and monotonic correlation coefficients
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04098-4
Robust linear and monotonic correlation measures capable of giving an accurate estimate of correlation when outliers are present, and reliable estimates when outliers are absent.
Based on the root mean square error (RMSE) and bias, the three proposed correlation measures are highly competitive when compared to classical measures such as Pearson and Spearman as well as robust measures such as Quadrant, Median, and Minimum Covariance Determinant.
□ VCFShark: how to squeeze a VCF file
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab211/6206359
VCFShark, which is able to compress VCF files up to an order of magnitude better than the de facto standards (gzipped VCF and BCF).
gPBWT (generalized positional Burrows–Wheeler transform) algorithm is a core of the GTShark algorithm. This is a different approach than used by genozip, which expands the genotypes in the whole chunk of VCF files to the largest ploidy present in this chunk.
□ Gene name errors: lessons not learned
>> https://www.biorxiv.org/content/10.1101/2021.03.30.437702v1.full.pdf

□ 4DNvestigator: Time Series Genomic Data Analysis Toolbox
>> https://www.tandfonline.com/doi/full/10.1080/19491034.2021.1910437
Data on genome organization and output over time, or the 4D Nucleome (4DN), require synthesis for meaningful interpretation. Development of tools for the efficient integration of these data is needed, especially for the time dimension.
4DNvestigator provide the definitions for multi-correlation and generalized singular values, the algorithm to compute tensor entropy, and an application of tensor entropy.
□ SynthDNM: Customized de novo mutation detection for any variant calling pipeline
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab225/6209072
SynthDNM, a random-forest based classifier that can be readily adapted to new sequencing or variant-calling pipelines by applying a flexible approach to constructing simulated training examples from real data.
The optimized SynthDNM classifiers predict de novo SNPs and indels with robust accuracy across multiple methods of variant calling.
□ AMICI: High-Performance Sensitivity Analysis for Large Ordinary Differential Equation Model
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab227/6209017
AMICI provides a multi-language (Python, C++, Matlab) interface for the SUNDIALS solvers CVODES (for ordinary differential equations) and IDAS (for algebraic differential equations). AMICI allows the user to read differential equation models specified as SBML or PySB.
As symbolic processing can be computationally intensive, AMICI symbolically only computes partial derivatives; total derivatives are computed through (sparse) matrix multiplication.
□ HCGA: highly comparative graph analysis for network phenotyping
>> https://www.cell.com/patterns/fulltext/S2666-3899(21)00041-6
The area closest in essence to HCGA is that of graph embeddings, in which the graph is reduced to a vector that aims to effectively incorporate the structural features.
the inherent choice of network properties that provide a “good” vector representation of the graph is not known and the type of statistical learning task. HCGA thus circumvents this critical step in the embedding process through indiscriminate massive feature extraction.










※コメント投稿者のブログIDはブログ作成者のみに通知されます