








問題の解決手段自体が、解決すべき問題になる。セグメントの単純化は、対象事物を指向するフラグメントの細分化を伴い、その複雑性を保存する。エントロピーは不可逆性を計る指標だが、複雑性は時間に対し可塑性を担保する。即ち行為の余波は、行為しなかった余波と対称的な力学量を持つ。
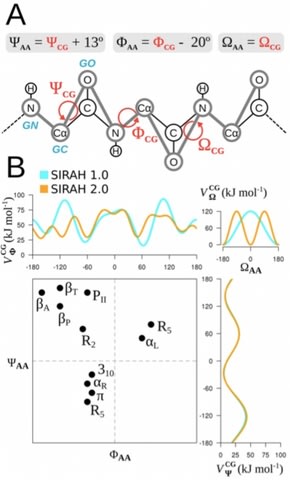
□ The SIRAH force field 2.0: Altius, Fortius, Citius:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/05/436774.full.pdf
SIRAH 2.0 can be considered a significant upgrade that comes at no increase of computational cost, as the functional form of the Hamiltonian, the number of beads in each moiety, and their topologies remained the same. The simulation of the holo form starting from an experimental structure sampled near- native conformations, retrieving quasi-atomistic precision.
□ A starless bias in the maximum likelihood phylogenetic methods:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/05/435412.full.pdf
If the aligned sequences are equidistant from each other with the true tree being a star tree, then the likelihood method is incapable of recover unless the sequences are either identical or extremely diverged. analyze this “starless” bias and identify the source for the bias. In contrast, distance-based methods (with the least-squares method for branch evaluation and either minimum evolution or least-squares criterion for choosing the best tree) do not have this bias. The finding sheds light on the star-tree paradox in Bayesian phylogenetic inference.
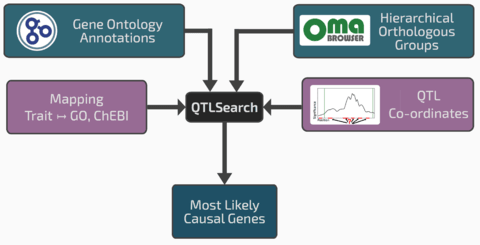
□ Prioritising candidate genes causing QTL using hierarchical orthologous groups:
>> https://academic.oup.com/bioinformatics/article/34/17/i612/5093215
Gene families, in the form of hierarchical orthologous groups from the Orthologous MAtrix project (OMA), enable reasoning over complex nested homologies in a consistent framework. By integrating functional inference with homology mapping, it is possible to differentiate the confidence in orthologous and paralogous relationships when propagating functional knowledge.
□ Evaluating stochastic seeding strategies in networks
>> https://arxiv.org/abs/1809.09561
how stochastic seeding strategies can be evaluated using existing data arising from randomized experiments in networks designed for other purposes and how to design much more efficient experiments for this specific evaluation. he proposed estimators and designs can dramatically increase precision while yielding valid inference.
□ CaSTLe – Classification of single cells by transfer learning: Harnessing the power of publicly available single cell RNA sequencing experiments to annotate new experiments:
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0205499
''CaSTLe–classification of single cells by transfer learning,'' is based on a robust feature engineering workflow and an XGBoost classification model built on these features. The feature engineering steps include: selecting genes with the top mean expression and mutual information gain, removing correlated genes, and binning the data according to pre-defined ranges.
□ Demonstration of End-to-End Automation of DNA Data Storage:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/10/439521.full.pdf
The device enables the encoding of data, which is then written to a DNA oligonucleotide using a custom DNA synthesizer, pooled for liquid storage, and read using a nanopore sequencer and a novel, minimal preparation protocol. The extension segment is then T/A ligated to the standard Oxford Nanopore Technology (ONT) LSK-108 kit sequencing adapter, creating the “extended ONT adapter,” which ensures that sufficient bases are read for successful base calling.

□ Selene: a PyTorch-based deep learning library for sequence-level data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/10/438291.full.pdf
"Sequence-level data" refers to any type of biological sequence such as DNA, RNA, or protein sequences and their measured properties (e.g. TF binding, DNase sensitivity, RBP binding). Training is automatically completed by Selene; afterwards, the researcher can easily use Selene to compare the performance of their new model to the original DeepSEA model on the same chromosomal holdout dataset.
□ DIAlign provides precise retention time alignment across distant runs in DIA and targeted proteomics:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/10/438309.full.pdf
DIAlign is a novel algorithm based on direct alignment of raw MS2 chromatograms using a hybrid dynamic programming approach. The algorithm does not impose a chronological order of elution and allows for aligning of elution-order swapped peaks.
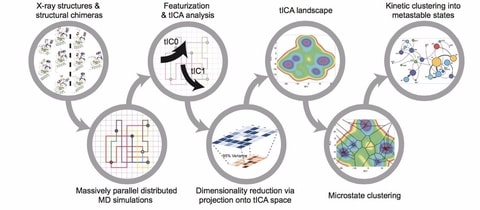
□ SETD8 wild-type apo and cofactor-bound, and mutant apo Folding@home simulations
>> https://osf.io/2h6p4/
□ VOMM: A framework for space-ecient variable-order Markov models:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/14/443101.full.pdf
a practical, versatile representations of variable-order Markov models and of interpolated Markov models, that support a large number of context-selection criteria, scoring functions, probability smoothing methods, and interpolations, and that take up to 4 times less space than previous implementations based on the suffix array, regardless of the number and length of contexts, and up to 10 times less space than previous trie-based representations, or more, while matching the size of related, state-of-the-art data structures from Natural Language Processing.
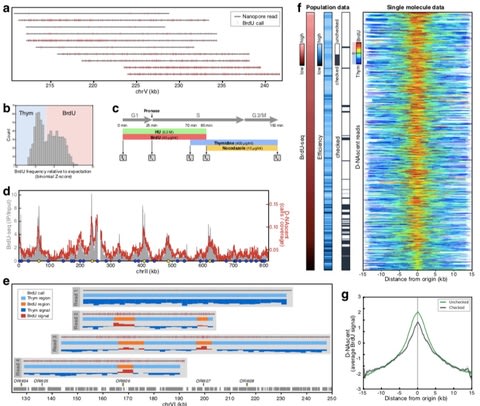
□ D-NAscent: Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/16/442814.full.pdf
Under conditions of limiting BrdU concentration, D-NAscent detects the differences in BrdU incorporation frequency across individual molecules to reveal the location of active replication origins, fork direction, termination sites, and fork pausing/stalling events. The trained BrdU pore model to account for the presence of BrdU in the sequence while also circumventing the high space and time complexities that can result from dynamic programming alignment. With an alignment of events to the Albacore basecall, then aligned to the minimap2.
□ Comparative Pathway Integrator: a framework of meta-analytic integration of multiple transcriptomic studies for consensual and differential pathway analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/16/444604.full.pdf
Given pathway enrichment results, perform Adaptively-weighted Fisher’s (AW Fisher) method as meta-analysis, to identify pathways significant in one or more studies/conditions.
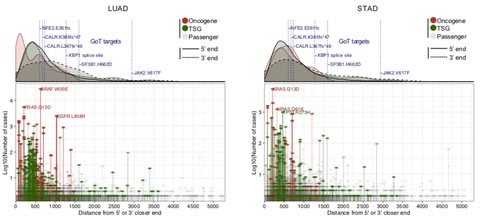
□ GoT: High throughput droplet single-cell Genotyping of Transcriptomes reveals the cell identity dependency of the impact of somatic mutations:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/16/444687.full.pdf
GoT capitalizes on high-throughput scRNA-seq (the 10x Genomics Chromium single cell 3’ platform), by which thousands of cells can be jointly profiled for genotyping information as well as single-cell full transcriptomes. the ability of GoT to genotype multiple target genes in parallel is critical. while described here for 3’ droplet-based scRNA-seq, GoT can be integrated in any scRNA-seq method that generates full length cDNA as an intermediate product (Microwell-seq, 10x SingleCell V(D)J +5′GE). the high-throughput linking of single-cell genotyping of expressed genes to transcriptomic data may provide the means to gain insight into questions such as the integration of clonal diversification with lineage plasticity or differentiation topologies.
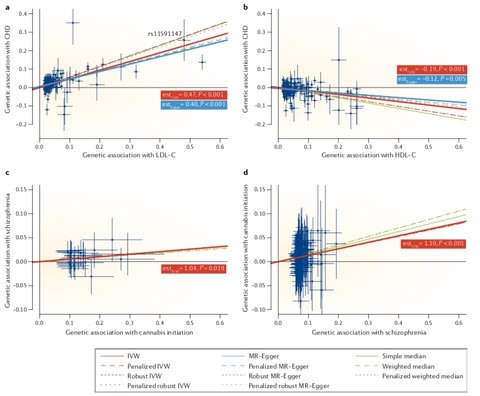
□ Using genetic data to strengthen causal inference in observational research:
>> https://www.nature.com/articles/s41576-018-0020-3
Recent progress in genetic epidemiology — including statistical innovation, massive genotyped data sets and novel computational tools for deep data mining — has fostered the intense development of methods exploiting genetic data and relatedness to strengthen causal inference. Assessing credibility requires in-depth knowledge of the question, which is unlikely in massive hypothesis-free causal inference exercises, such as phenome-wide approaches.
Triangulation — when conclusions from several study designs converge — will play an increasingly important role in strengthening evidence for causality. One should not expect that a single existing or future method for causal inference in observational settings will provide a definitive answer to a causal question. Rather, such methods can substantially improve the strength of evidence on a continuum from mere association to established causality.
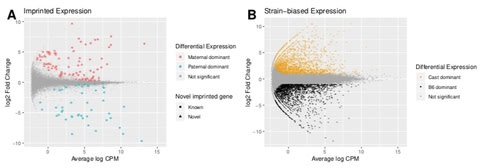
□ Using long-read sequencing to detect imprinted DNA methylation:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/17/445924.full.pdf
Determining allele-specific methylation patterns in diploid or polyploid cells with short-read sequencing is hampered by the dependence on a high SNP density and the reduction in sequence complexity inherent to bisulfite treatment. Using long-read nanopore sequencing, with an average genomic coverage of approximately ten, it is possible to determine both the level of methylation of CpG sites and the haplotype from which each read arises.
□ BiG-SCAPE and CORASON: A computational framework for systematic exploration of biosynthetic diversity from large-scale genomic data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/17/445270.full.pdf
BiG-SCAPE facilitates rapid calculation and interactive exploration of BGC sequence similarity networks (SSNs); it accounts for differences in modes of evolution between BGC classes, groups gene clusters at multiple hierarchical levels, introduces a ‘glocal’ alignment mode that supports complete as well as fragmented BGCs, and democratizes the analysis through a dramatically accelerated.
□ Reverse GWAS: Using Genetics to Identify and Model Phenotypic Subtypes:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/17/446492.full.pdf
Reverse GWAS (RGWAS) to identify and validate subtypes using genetics and multiple traits: while GWAS seeks the genetic basis of a given trait, RGWAS seeks to define trait subtypes with distinct genetic bases. RGWAS uses a bespoke decomposition, MFMR, to model covariates, binary traits, and population structure. A random-effect version of MFMR could improve power to detect polygenic subtypes, though computational issues are non-trivial. MFMR could also be adapted to count data, zero-inflation, higher-order arrays, or missing data.
□ OSCA: a tool for omic-data-based complex trait analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/17/445163.full.pdf
MOMENT (a mixed-linear-model-based method) that tests for association between a DNAm probe and trait with all other distal probes fitted in multiple random-effect components to account for the effects of unobserved confounders as well as the correlations between distal probes. MOMENT has been implemented in a versatile software package (OSCA) together with a number of other implementations for omic-data-based analysis incl the estimation of variance in a trait captured by all measures of multiple omic profiles, xQTL analysis, and meta-analysis of xQTL.

□ LuxRep: a technical replicate-aware method for bisulfite sequencing data analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/19/444711.full.pdf
LuxGLM is a probabilistic covariate model for quantification of DNA methylation modifications with complex experimental designs. LuxRep improves the accuracy of differential methylation analysis and lowers running time of model-based DNA methylation analysis. LuxRep features Model-based integration of biological / technical replicates, and Full Bayesian inference by variational inference implemented in Stan. LuxRep also generates count data from sequencing reads using e.g. Bismark, and align BS-seq data.
□ RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/18/447110.full.pdf
RAxML-NG is a from scratch re-implementation of the established greedy tree search algorithm of RAxML/ExaML. RAxML and ExaML are large monolithic codes, RAxML-NG employs a two-step L-BFGS-B method to optimize the parameters of the LG4X model. RAxML-NG is a phylogenetic tree inference tool which uses maximum-likelihood (ML) optimality criterion. RAxML can compute the novel branch support metric called transfer bootstrap expectation. TBE is less sensitive to individual misplaced taxa in replicate trees, and thus better suited to reveal well-supported deep splits in large trees with thousands of taxa.
□ On Parameter Interpretability of Phenomenological-Based Semiphysical Models:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/18/446583.full.pdf
the phenomenological modeling approach offers the great advantage of having a structure with variables and parameters with physical meaning that enhance the interpretability of the model and its further used for decision making. this property has not been deeply discussed, perhaps by the implicit assumption that interpretability is inherent to the phenomenological-based models.
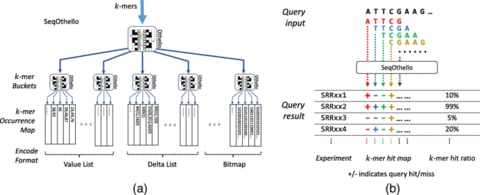
□ SeqOthello: querying RNA-seq experiments at scale:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1535-9
SeqOthello, an ultra-fast & memory-efficient indexing structure to support arbitrary sequence query against large collections of RNA-seq experiments. It takes only 5 min and 19.1 GB memory to conduct a global survey of 11,658 fusion events against 10,113 TCGA Pan-Cancer datasets. By providing a reference-free, alignment-free, and parameter-free sequence search system, SeqOthello will enable large-scale integrative studies using sequence-level data, an undertaking not previously practicable for many individual labs.

□ Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures:
>> https://www.nature.com/articles/nbt.4278
The obtained sparse fluorescent sequence of each molecule was then assigned to its parent protein in a reference database. testing the method on synthetic and naturally derived peptide molecules in zeptomole-scale quantities. they also fluorescently labeled phosphoserines and achieved single-molecule positional readout of the phosphorylated sites.
□ Deciphering epigenomic code for cell differentiation using deep learning:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/22/449371.full.pdf
Increasing lines of evidence have suggested that the epigenome in a cell type is established step-wisely though the interplay of genomic sequence, chromatin remodeling systems and environmental cues along the developmental lineage. As the latter two factors are the results of interactions of the products of genomic sequences, the epigenome of a cell type is ultimately determined by the genomic sequence.
□ A relative comparison between Hidden Markov- and Log-Linear- based models for differential expression analysis in a real time course RNA sequencing data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/22/448886.full.pdf
evaluate the relative performance of two Hidden Markov- and Log-Linear- based statistical models in detection of DE genes in a real time course RNA-seq data. The Hidden Markov-based model, EBSeq-HMM, was particularly developed for time course experiments while the log-linear based model, multiDE, was proposed for multiple treatment conditions.
□ Efficient Proximal Gradient Algorithm for Inference of Differential Gene Networks:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/22/450130.full.pdf
The differential gene-gene interactions identified by ProGAdNet algorithm yield a list of genes alternative to the list of differentially expressed genes. This may provide additional insight into the molecular mechanism behind the phenotypical difference of the tissue under different conditions. Alternatively, the two gene networks inferred by ProGAdNet algorithm can be used for further differential network analysis (DiNA).
□ Look4TRs: A de-novo tool for detecting simple tandem repeats using self-supervised hidden Markov models:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/23/449801.full.pdf
Look4TRs adapts itself to the input genomes automatically, balancing high sensitivity and low false positive rate. Look4TRs generates a random chromosome based on a real chromosome of the input genome. Then it inserts semi-synthetic MS in the random chromosome. Finally, the HMM is trained and calibrated using these semi-synthetic MS.










※コメント投稿者のブログIDはブログ作成者のみに通知されます