










□ scPhere: Deep generative model embedding of single-cell RNA-Seq profiles on hyperspheres and hyperbolic spaces
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/25/853457.full.pdf
For general scRNA-seq data, scPhere minimize the distortion by embedding cells to a lower-dimensional hypersphere instead of a low-dimensional Euclidean space, using von Mises-Fisher (vMF) distributions on hyperspheres as the posteriors for the latent variables.
By using a hyperspherical latent space, scPhere overcomes the problem of forcing cells to center at the origin of the latent space.
ScPhere resolves cell crowding, corrects multiple, complex batch factors, facilitates interactive visualization of large datasets, and gracefully uncovers pseudotemporal trajectories.

□ PHATE: Visualizing structure and transitions in high-dimensional biological data
>> https://www.nature.com/articles/s41587-019-0336-3
define a manifold preservation metric, which called denoised embedding manifold preservation (DEMaP), and PHATE produces lower-dimensional embeddings that are quantitatively better denoised as compared to existing visualization methods.
PHATE (Potential of Heat-diffusion for Affinity-based Trajectory Embedding) captures both local and global nonlinear structure using an information-geometric distance between data points.
PHATE creates a diffusion-potential geometry by free-energy potentials of these processes. This geometry captures high-dimensional trajectory structures, while enabling a natural embedding of the intrinsic data geometry.


□ Clustering-independent analysis of genomic data using spectral simplicial theory
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007509
Combinatorial Laplacian scores take into account the topology spanned by the data and reduce to the ordinary Laplacian score when restricted to graphs.
the utility of this framework with several applications to the analysis of gene expression and multi-modal genomic data, it provides a unifying perspective on topological data analysis and manifold learning approaches to the analysis of large-scale biological datasets.

□ CORE GREML: Estimating covariance between random effects in linear mixed models for genomic analyses of complex traits
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/25/853515.full.pdf
the covariance between random effects is a key parameter that needs to be estimated, especially when partitioning phenotypic variance by omic layer.
CORE GREML is useful for genomic partitioning analyses and for genome-transcriptome partitioning of phenotypic variance.
CORE GREML (CORE for COvariance between Random Effects), that fits the Cholesky decomposition of kernel matrices in a LMM to estimate the covariance between a given pair of random effects.

□ Detecting, Categorizing, and Correcting Coverage Anomalies of RNA-Seq Quantification
>> https://www.cell.com/cell-systems/fulltext/S2405-4712(19)30381-3
SAD detects quantification anomalies using the disagreement between the modeled expected coverage and the observed fragment coverage distribution that is obtained after the quantifier has allocated fragments to transcripts.
Anomaly categorization is done by reassigning the reads across the isoforms using linear programming and checking whether the anomaly score becomes insignificant after the fragment reassignment.

□ Strand-seq: A fully phased accurate assembly of an individual human genome
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/26/855049.full.pdf
Using Strand-seq data and SaaRclust, they readily corrected contig misorientations and chimerisms in the Peregrine assembly and found the majority (~76%) of these misassemblies overlapped or mapped in the vicinity of SDs of size 50 kbp and longer.
A comparison of Oxford Nanopore and PacBio phased assemblies identifies 150 regions that are preferential sites of contig breaks irrespective of sequencing technology or phasing algorithms.
Using Strand-seq’s capacity to preserve structural and directional contiguity of individual homologs, it assigns phased Peregrine contigs into whole chromosomal scaffolds, again using the process described above for the collapsed assemblies.

□ Wengan: Efficient and high quality hybrid de novo assembly of human genomes
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/25/840447.full.pdf
Wengan is a new genome assembler that unlike most of the current long-reads assemblers avoids entirely the all-vs-all read comparison.
The key idea behind Wengan is that long-read alignments can be inferred by building paths on a sequence graph.
Wengan builds a new sequence graph called the Synthetic Scaffolding Graph. The SSG is built from a spectrum of synthetic mate-pair libraries extracted from raw long-reads. Longer alignments are then built by peforming a transitive reduction of the edges.

□ A New approximate matching compression algorithm for DNA sequences
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/25/853358.full.pdf
Calculating the relative and total frequencies given by the permutation formula (nr) and compressing 6 bits of information into 1 implementing the ASCII table code (0...255 characters, 28), using clusters of 102 DNA bases compacted into 17 string sequences.
For decompressing, the inverse process must be done, except that the triplets must be selected randomly (or use a matrix dictionary, 4102). The compression algorithm has a better compression ratio than LZW and Huffman's algorithm.

□ CAPITAL: Alignment of time-course single-cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/11/29/859751.full.pdf
CAPITAL, a method for comparing pseudotime trajectories with tree alignment whereby trajectories including branching can be compared without any knowledge of paths to be compared.
Computational tests on time-series public data indicate that CAPITAL can align non-linear pseudotime trajectories and reveal gene expression dynamics.
□ Phylonium: Fast Estimation of Evolutionary Distances from Large Samples of Similar Genomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz903/5650408
Phylonium is a faster version of the published program andi. phylonium is particularly fast when applied to large samples and is more accurate than mash when applied to sequences where homology is only local.
Phylonium is based on the same anchor distances as andi, the accuracy of the two programs is similarly high while Phylonium is much faster than alignment based approaches for phylogeny reconstruction and usually more accurate than competing alignment-free methods.
□ Minipolish: A tool for Racon polishing of miniasm assemblies
>> https://github.com/rrwick/Minipolish
Minipolish assumes that you have minimap2 and Racon installed and available in your PATH. With a single command, it will use Racon to polish up a miniasm assembly, while keeping the assembly in graph form.
Minipolish finishes by doing one more read-to-assembly alignment, this time not to polish but to calculate read depths. These depths are added to the Graphical Fragment Assembly (GFA) line for each contig.

□ QAlign: Aligning nanopore reads accurately using current-level modeling
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/03/862813.full.pdf
QAlign, a pre-processor that can be used with any long-read aligner for aligning long reads to a genome / transcriptome or to other long reads.
The key idea in QAlign is to convert the nucleobase reads into discretized current levels that capture the error modes of the nanopore sequencer before running it through a sequence aligner.

□ DeePlexiCon: Barcoding and demultiplexing Oxford Nanopore native RNA sequencing reads with deep residual learning
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/04/864322.full.pdf
a novel strategy to barcode and demultiplex direct RNA sequencing nanopore data, which does not rely on base-calling or additional library preparation steps.
DeePlexiCon, implements a 20-layer residual neural network model that can demultiplex 93% of the reads with 95.1% specificity, or 60% of reads with 99.9% specificity.
Demultiplexing is performed via the transformation of raw FAST5 signals into images using Gramian Angular Summation Field (GASF), followed by classification using a deep residual neural network learning model.

□ WEDGE: recovery of gene expression values for sparse single-cell RNA-seq datasets using matrix decomposition
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/04/864488.full.pdf
a new algorithm, WEDGE (WEighted Decomposition of Gene Expression), which imputes expression matrix by using a low-rank matrix decomposition method and a low weight parameter for the zero elements in the expression matrix.
In WEDGE, adopting a lower weight for the zero elements in the expression matrix during the low-rank decomposition, and generated a convergent recovered matrix by the alternating non-negative least-squares algorithm.

□ DENs: Deep exploration networks for rapid engineering of functional DNA sequences
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/04/864363.full.pdf
deep exploration networks (DENs), a type of generative model tailor-made for searching a sequence space to minimize the cost of a neural network fitness predictor.
The generator produces sequence patterns which the predictor evaluates on the basis of an objective function; the overall goal is to generate sequences maximizing the objective.

□ Afann: bias adjustment for alignment-free sequence comparison based on sequencing data using neural network regression
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1872-3
The alignment-free dissimilarity calculated based on sequencing samples can be overestimated compared with the dissimilarity calculated based on their genomes, and this bias can significantly decrease the performance of the alignment-free analysis.
Afann (Alignment-Free methods Adjusted by Neural Network) is an alignment-free software that supports fast calculation of different dissimilarity measures including d2star, d2shepp, CVtree, Manhattan, Euclidean and d2 without downsampling or estimating the sequencing depth.
□ Sierra: Discovery of differential transcript usage from polyA-captured single-cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/07/867309.full.pdf
Sierra detects genomic regions corresponding to potential polyA sites enables a data-driven approach to detecting novel DTU events, such as alternative 3’ usage and APA between single-cell populations.
Sierra is applicable to barcoded droplet-based scRNA-seq experimental data such as produced using the 10x Genomics Chromium system.
After finding the exonic peaks, Sierra goes back to each splice junction and try to fit a Gaussian curve to the “intronic” peak coverage within the splice junction. Sierra ignores peaks in intergenic regions.

□ ACE: A Probabilistic Model for Characterizing Gene-Level Essentiality in CRISPR Screens
>> https://www.biorxiv.org/content/biorxiv/early/2019/12/08/868919.full.pdf
ACE considers each experimental phase of the CRISPR-Cas9 screen — the master library infection, the initial and final sequencing — as a separate probabilistic process forming a hierarchical Bayesian model, simulated datasets provide a reliable ground truth.
The core of ACE model is a series of Poisson sampling processes integrated over true sgRNA abundance, with a likelihood ratio test to evaluate significance. Essentiality estimates φG ∈ [−∞,1] are calculated for every genetic element G.
□ wtdbg2: Fast and accurate long-read assembly
>> https://www.nature.com/articles/s41592-019-0669-3
Existing long-read assemblers require thousands of central processing unit hours to assemble a human genome and are being outpaced by sequencing.
a long-read assembler wtdbg2 that is 2–17 times as fast as published tools while achieving comparable contiguity and accuracy.
Wtdbg2 is able to assemble the human and even the 32Gb Axolotl genome at a speed tens of times faster than CANU and FALCON while producing contigs of comparable base accuracy.
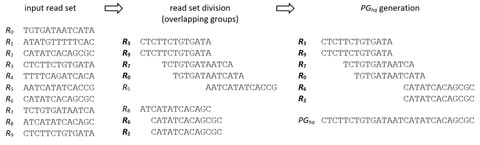
□ PgRC: Pseudogenome based Read Compressor
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz919/5670526
Pseudogenome-based Read Compressor (PgRC), an in-memory algorithm for compressing the DNA stream, based on the idea of building an approximation of the shortest common superstring over high-quality reads.
PgRC wins in compression ratio over its main competitors, SPRING and Minicom, by up to 15 and 20 percent on average, respectively, while being comparably fast in decompression.
□ A DNA-of-things storage architecture to create materials with embedded memory
>> https://www.nature.com/articles/s41587-019-0356-z
□ LIBRA-seq: High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity
>> https://www.cell.com/cell/pdf/S0092-8674(19)31224-3.pdf
□ Miodin: Vertical and horizontal integration of multi-omics data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3224-4
Miodin allows users to integrate omics data from different experiments on the same samples (vertical integration) or across studies on the same variables (horizontal integration).
A key design goal when developing miodin was to streamline data analysis, by providing a clean syntax for building workflows and minimizing the extent of technical expertise required to combine multiple software packages.
□ Arteria: An automation system for a sequencing core facility
>> https://academic.oup.com/gigascience/article/8/12/giz135/5673459
Arteria can be reduced to 3 conceptual levels: orchestration (using an event-based model of automation), process (the steps involved in processing sequencing data, modelled as workflows), and execution (using a series of RESTful micro-services).
Arteria uses the open source automation platform StackStorm for event-based orchestration and the Mistral workflow engine.

□ Lean and Deep Models for More Accurate Filtering of SNP and INDEL Variant Calls
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz901/5674040
Bayesian hyper-parameter optimization confirmed this notion that architectures designed with DNA data in mind outperform off-the-shelf image classification models.
The cross-generalization analysis identified idiosyncrasies in truth resources pointing to the need for new methods to construct genomic truth data.

□ GABAC: an arithmetic coding solution for genomic data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz922/5674036
GABAC combines proven coding technologies, such as context-adaptive binary arithmetic coding, binarization schemes and transformations, into a straightforward solution for the compression of sequencing data.
GABAC, the first implementation of an entropy codec outperforms well-established (entropy) codecs in a significant set of cases and thus can serve as an extension for existing genomic compression solutions, such as CRAM.
□ Circle-Map: Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3160-3
Circle-Map takes as input an alignment of reads to a reference genome (e.g. a BWA-MEM generated BAM file), it will use those alignments to detect cases were the read has been split into two segments to detect genomic rearrangements supporting a circular DNA structure.
Circle-Map guides the realignment of partially aligned reads using information from discordantly mapped reads to map the short unaligned portions using a probabilistic model.
□ Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1865-2
Vireo (Variational Inference for Reconstructing Ensemble Origins), a computationally efficient Bayesian model to demultiplex single-cell data from pooled experimental designs.
Uniquely, Vireo can be applied in settings when only partial or no genotype information is available. Vireo models the genotypes of each individual as latent variables, which are inferred from the observed scRNA-seq reads. The model can also leverage partial genotype information.

□ K-Phylo: An Alignment-free Method for Phylogeny Estimation using Maximum Likelihood
>> https://www.biorxiv.org/content/10.1101/2019.12.13.875526v1.full.pdf
K-Phylo, an alignment-free method for phylogeny reconstruction that uses maximum likelihood for tree estimation.
K-Phylo is based on a simple alignment free method for phylogeny construction based on contiguous sub-sequences of length k termed k-mers.

□ Finding ranges of optimal transcript expression quantification in cases of non-identifiability
>> https://www.biorxiv.org/content/10.1101/2019.12.13.875625v1.full.pdf
Analyzing the overlap of ranges of optima in differential expression (DE) detection reveals that the majority of predictions are reliable, but there are a few unreliable predictions for which switching to other optimal abundances may lead to similar expression between DE conditions.
Assuming that the reference transcriptomis incomplete but the splice graph is correct, proposing a set of max-flow-based algorithms on an adapted splice graph for reconstructing the set of optima.
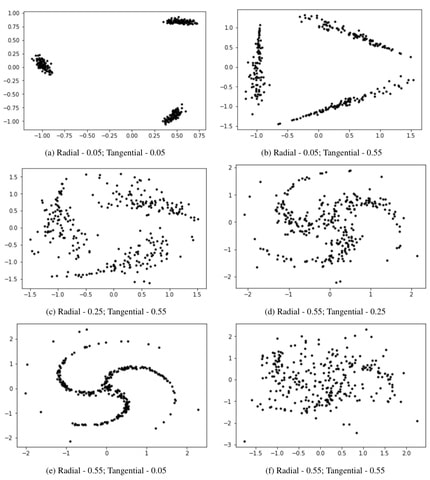
□ Clustering using Automatic Differentiation: Misspecified models and High-Dimensional Data
>> https://www.biorxiv.org/content/10.1101/2019.12.13.876326v1.full.pdf
These empirical analyses reveal limitations of likelihood based model selection in both Expectation Maximiza- tion (EM) and Gradient Descent (GD) based inference methods.
Design new KL divergence based model selection criteria and GD-based in- ference methods that use the criteria in fitting GMM on low- and high-dimensional data as well as for selecting the number of clusters.
This criterion is used along w/ likelihood as an objective function for GMM parameter estimation for clustering high- dimensional data. A suitable variant, called Maximum absolute Pairwise difference between KL divergence (MPKL) is designed for model selection in high dimensions.
□ MPF-BML: A standalone GUI-based package for maximum entropy model inference https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz925/5680343
a statistical method, Minimum Probability Flow-Boltzmann Machine Learning (MPF-BML), for performing fast and accurate inference of maximum entropy model parameters, which was applied to genetic sequence data.
Maximum epsilon value for the individual, pairwise and connected correlations. The epsilon value is chosen to balance between underfitting and overfitting the individual - pairwise - connected correlations. Higher value will mean less accuracy but faster convergence.

□ Accurate and simultaneous identification of differential expression and splicing using hierarchical Bayesian analysis
>> https://www.biorxiv.org/content/10.1101/2019.12.16.878884v1.full.pdf
Hierarchical Bayesian Analysis of Differential Expression and ALternative SPlicing (HBA-DEALS) consists of a matrix of gene and isoform counts derived from to two different conditions, here referred to as case and control, using any short- or long-read next- generation sequencing technology.
The algorithmic approach HBA-DEALS is a paradigm that can be used to determine the existence of DAST, DGE, both, or neither.

□ OnTAD: hierarchical domain structure reveals the divergence of activity among TADs and boundaries
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1893-y
OnTAD reveals new biological insights into the role of different TAD levels, boundary usage in gene regulation, the loop extrusion model, and compartmental domains.
the candidate boundaries are assembled into the optimized hierarchical TAD structures using a recursive dynamic programming algorithm based on a scoring function.
□ Fully moderated t-statistic in linear modeling of mixed effects for differential expression analysis
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3248-9
an approach to allow variance shrinkage of both residual error and random effects using moderated t-statistic under LMM.
the implementation of the fully moderated t-statistic method for linear models with mixed effects, where both residual variances and variance estimates of random effects are smoothed under a hierarchical Bayes framework.

□ Paragraph: a graph-based structural variant genotyper for short-read sequence data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1909-7
Paragraph works by aligning and genotyping reads on a local sequence graph constructed for each targeted SV. This approach is different from other proposed and most existing graph methods that create a single whole-genome graph and align all reads to this large graph.
Demonstrating the accuracy of Paragraph on whole-genome sequence data from three samples using long-read SV calls as the truth set, and then apply Paragraph at scale to a cohort of 100 short-read sequenced samples of diverse ancestry.
□ StringTie2: Transcriptome assembly from long-read RNA-seq alignments
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1910-1
StringTie2 includes new methods to handle the high error rate of long reads and offers the ability to work with full-length super-reads assembled from short reads, which further improves the quality of short-read assemblies.
When applied to long reads, StringTie2 assembles the reads substantially more accurately, faster, and using less memory than FLAIR, the next-best performing tool for long-read analysis.

□ LPM: a latent probit model to characterize the relationship among complex traits using summary statistics from multiple GWASs and functional annotations
>>https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz947/5682406
a latent probit model (LPM) to integrate summary-level GWAS data and functional annotations. The developed computational framework not only makes LPM scalable to hundreds of annotations and phenotypes, but also ensures its statistically guaranteed accuracy.
To make LPM scalable to millions of SNPs and hundreds of traits, instead of working with a brute-force algorithm to handle all the data simultaneously, developed an efficient PX-EM algorithm and implement a dynamic threading strategy to enhance its parallel property.

□ MethylStar: A fast and robust pipeline for high-throughput analysis of bulk or single-cell WGBS data
>> https://www.biorxiv.org/content/10.1101/2019.12.20.884536v1.full.pdf
MethylStar integrates processing of raw fastq reads for both single- and paired-end data with options for adapter trimming (Trimmomatic), quality control (fastQC) and removal of PCR duplicates (Bismark).










※コメント投稿者のブログIDはブログ作成者のみに通知されます