









(Created with Midjourney v6 ALPHA)
□ Aaron Hibell / “Oblivion”
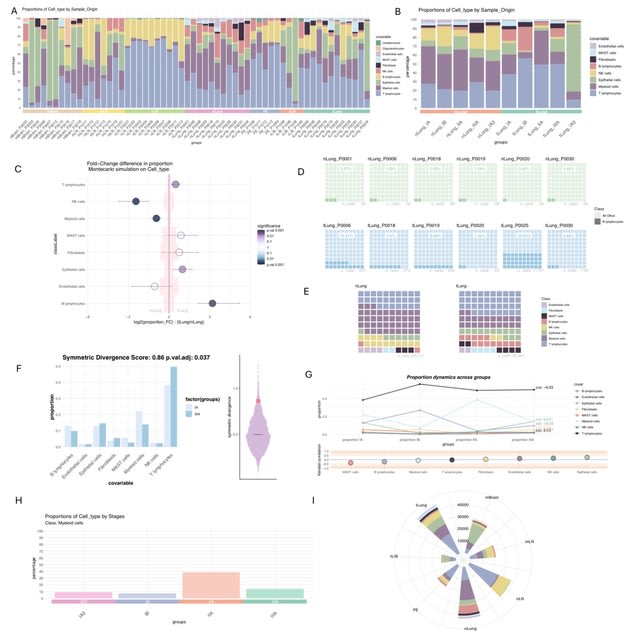
□ LotOfCells: data visualization and statistics of single cell metadata
>> https://www.biorxiv.org/content/10.1101/2024.05.23.595582v1
LotOfCells, an R package to easily visualize and analyze the phenotype data (metadata) from single cell studies. It allows to test whether the proportion of the number of cells from a specific population is significantly different due to a condition or covariate.
LotOfCells introduces a symmetric score, based on the Kullback-Leibler (KL) divergence, a measure of relative entropy between probability distributions.
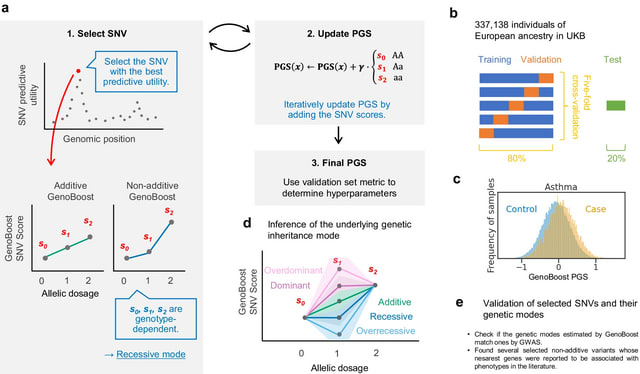
□ GenoBoost: A polygenic score method boosted by non-additive models
>> https://www.nature.com/articles/s41467-024-48654-x
GenoBoost, a flexible PGS modeling framework capable of considering both additive and non-additive effects, specifically focusing on genetic dominance. The GenoBoost algorithm fits a polygenic score (PGS) function in an iterative procedure.
GenoBoost selects the most informative SNV for trait prediction conditioned on the previously characterized effects and characterizes the genotype-dependent scores. GenoBoost iteratively updates its model using two hyperparameters: learning rate γ and the number of iterations.
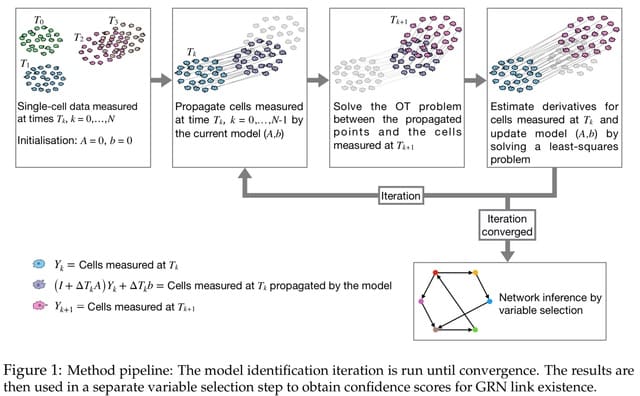
□ GRIT: Gene regulatory network inference from single-cell data using optimal transport
>> https://www.biorxiv.org/content/10.1101/2024.05.24.595731v1
GRIT, a method based on fitting a linear differential equation model. GRIT works by propagating cells measured at a certain time, and calculating the transport cost between the propagated population and the cell population measured at the next time point.
GRIT is essentially a system identification tool for linear discrete-time systems from population snapshot data. To investigate the performance of the method in this task, it is here applied on data generated from a 10-dimensional linear discrete-time system.
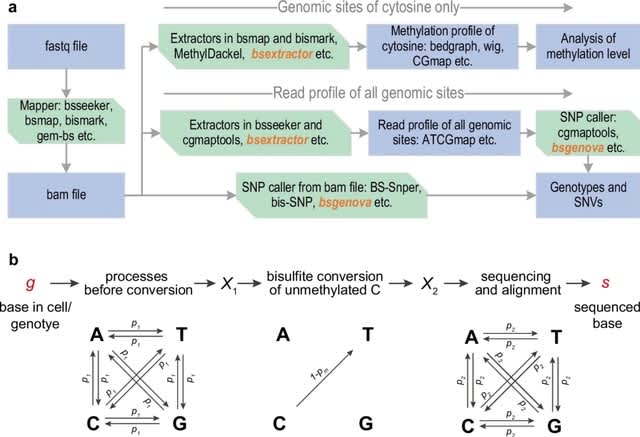
□ bsgenova: an accurate, robust, and fast genotype caller for bisulfite-sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05821-7
bsgenova, a novel SNP caller tailored for bisulfite sequencing data, employing a Bayesian multinomial model. Bsgenova uses a summary ATCGmap file as input which incl. the essential reference base, CG context, and ATCG read counts mapped onto Watson and Crick strands respectively.
bsgenova builds a Bayesian probabilistic model of read counts for each specific genomic position to calculate the (posterior) probability of a SNP.
In addition to utilizing matrix computation, bsgenova incorporates multi-process parallelization for acceleration. bsgenova reads data from file or pipe and maintains an in-memory cache pool of data batches of genome intervals.

□ GraphAny: A Foundation Model for Node Classification on Any Graph
>> https://arxiv.org/abs/2405.20445
GraphAny consists of two components: a LinearGNN that performs inference on new feature and label spaces without training steps, and an attention vector for each node based on entropy-normalized distance features that ensure generalization to new graphs.
GraphAny employs multiple LinearGNN models with different graph convolution operators and learn an attention vector. GraphAny enables entropy normalization to rectify the distance feature distribution to a fixed entropy, which reduces the effect of different label dimensions.

□ ProCapNet: Dissecting the cis-regulatory syntax of transcription initiation with deep learning
>> https://www.biorxiv.org/content/10.1101/2024.05.28.596138v1
ProCapNet accurately models base-resolution initiation profiles from PRO-cap experiments using local DNA sequence.
ProCapNet learns sequence motifs with distinct effects on initiation rates and TSS positioning and uncovers context-specific cryptic initiator elements intertwined within other TF motifs.
ProCapNet annotates predictive motifs in nearly all actively transcribed regulatory elements across multiple cell-lines, revealing a shared cis-regulatory logic across promoters and enhancers mediated by a highly epistatic sequence syntax of cooperative motif interactions.
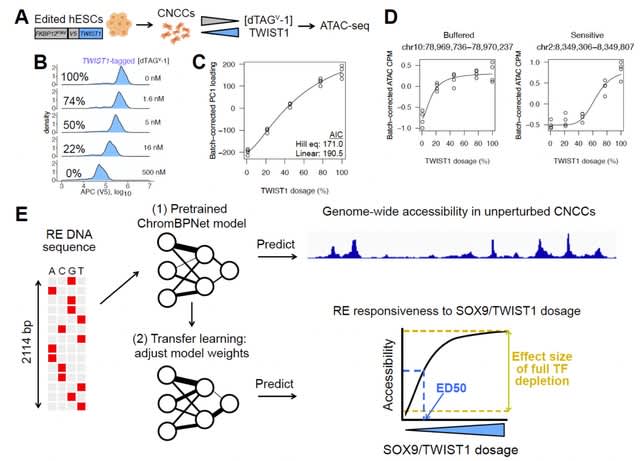
□ Transfer learning reveals sequence determinants of the quantitative response to transcription factor dosage
>> https://www.biorxiv.org/content/10.1101/2024.05.28.596078v1
Combining transfer learning of chromatin accessibility models with TF dosage titration by dTAG to learn the sequence logic underlying responsiveness to SOX9 and TWIST1 dosage in CNCCs.
This approach predicted how REs responded to TF dosage, both in terms of magnitude and shape of the response (sensitive or buffered), with accuracy greater than baseline methods and approaching experimental reproducibility.
Model interpretation revealed both a TF-shared sequence logic, where composite or discrete motifs allowing for heterotypic TF interactions predict buffered responses, and a TF-specific logic, where low-affinity binding sites for TWIST1 predict sensitive responses.
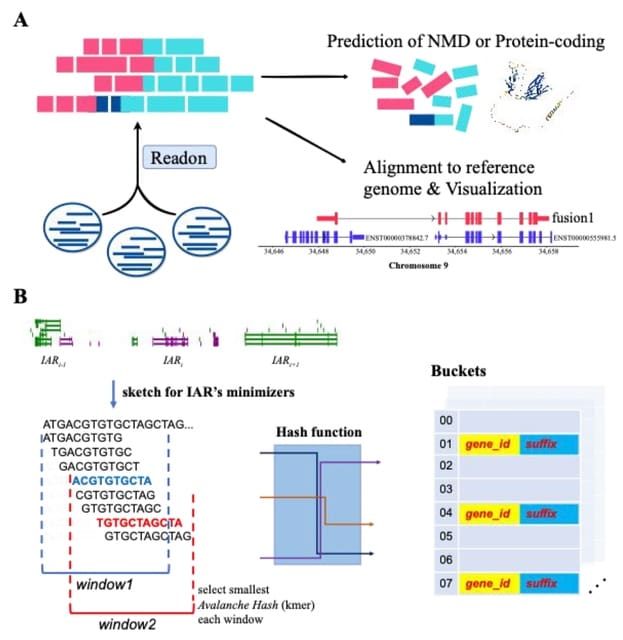
□ Readon: a novel algorithm to identify read-through transcripts with long-read sequencing data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae336/7684264
Readon, a novel minimizer sketch algorithm which effectively utilizes the neighboring position information of upstream and downstream genes by isolating the genome into distinct active regions.
Readon employs a sliding window within each region, calculates the minimizer and builds a specialized, query-efficient data structure to store minimizers. Readon enables rapid screening of numerous sequences that are less likely to be detected as read-through transcripts.
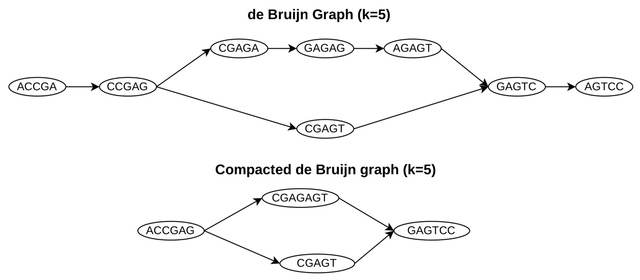
□ Cdbgtricks: strategies to update a compacted de bruijn graph
>> https://www.biorxiv.org/content/10.1101/2024.05.24.595676v1
Cdbgtricks, a novel strategy, and a method to add sequences to an existing uncolored compacted de Bruin graph. Cdbgtricks takes advantage of kmtricks that finds in a fast way what k-mers are to be added to the graph.
Cdbgtricks enables us to determine the part of the graph to be modified while computing the unitigs from these k-mers. The index of Cdbgtricks is also able to report exact matches between query reads and the graph. Cdbgtricks is faster than Bifrost and GGCAT.
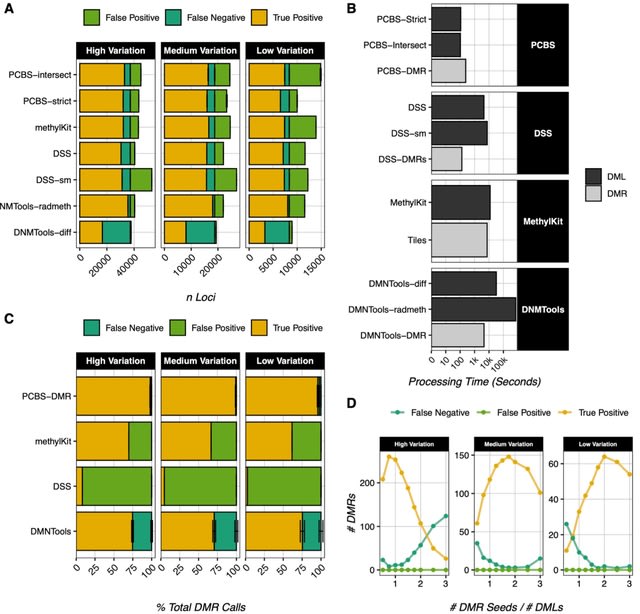
□ PCBS: an R package for fast and accurate analysis of bisulfite sequencing data
>> https://www.biorxiv.org/content/10.1101/2024.05.23.595620v1
PCBS (Principal Component BiSulfite) a novel, user-friendly, and computationally-efficient R package for analyzing WGBS data holistically. PCBS is built on the simple premise that if a PCA strongly delineates samples between two conditions.
Then the value of a methylated locus in the eigenvector of the delineating principal component (PC) will be larger if that locus is highly different between conditions.
Thus, eigenvector values, which can be calculated quickly for even a very large number of sites, can be used as a score that roughly defines how much any given locus contributes to the variation between two conditions.
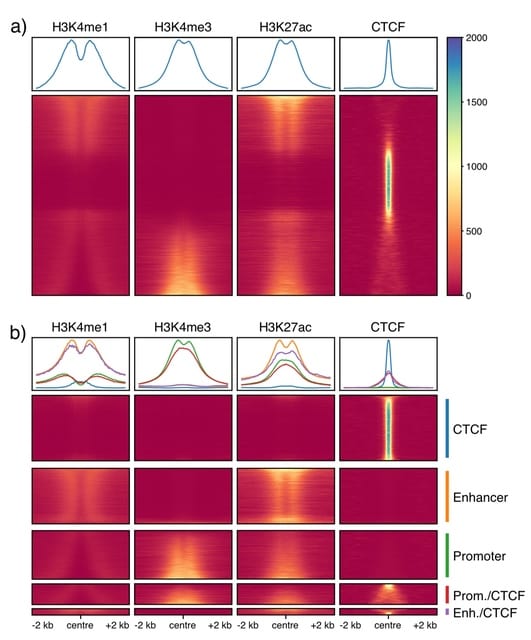
□ Deciphering cis-regulatory elements using REgulamentary
>> https://www.biorxiv.org/content/10.1101/2024.05.24.595662v1
REgulamentary, a standalone, rule-based bioinformatic tool for the thorough annotation of cis-regulatory elements for chromatin-accessible or CTCF-binding regions of interest.
REgulamentary is able to correctly identify this feature due to the correct ranking of the relative signal strength of the two chromatin marks.
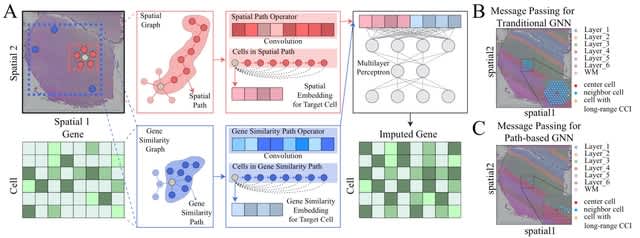
□ Impeller: a path-based heterogeneous graph learning method for spatial transcriptomic data imputation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae339/7684233
Impeller, a path-based heterogeneous graph learning method for spatial transcriptomic data imputation. Impeller builds a heterogeneous graph with two types of edges representing spatial proximity and expression similarity.
Impeller can simultaneously model smooth gene expression changes across spatial dimensions and capture similar gene expression signatures of faraway cells from the same type.
Impeller incorporates both short- and long-range cell-to-cell interactions (e.g., via paracrine and endocrine) by stacking multiple GNN layers. Impeller uses a learnable path operator to avoid the over-smoothing issue of the traditional Laplacian matrices.

□ Pantry: Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics
>> https://www.biorxiv.org/content/10.1101/2024.05.14.594051v1
Pantry (Pan-transcriptomic phenotyping), a framework to efficiently generate diverse RNA phenotypes from RNA-seq data and perform downstream integrative analyses with genetic data.
Pantry currently generates phenotypes from six modalities of transcriptional regulation (gene expression, isoform ratios, splice junction usage, alternative TSS/polyA usage, and RNA stability) and integrates them w/ genetic data via QTL mapping, TWAS, and colocalization testing.

□ GRanges: A Rust Library for Genomic Range Data
>> https://www.biorxiv.org/content/10.1101/2024.05.24.595786v1
GRanges, a Rust-based genomic ranges library and command-line tool for working with genomic range data. The goal of GRanges is to strike a balance between the expressive grammar of plyranges, and the performance of tools written in compiled languages.
The GRanges library has a simple yet powerful grammar for manipulating genomic range data that is tailored for the Rust language's ownership model. Like plyranges and tidyverse, the GRanges library develops its own grammar around an overlaps-map-combine pattern.
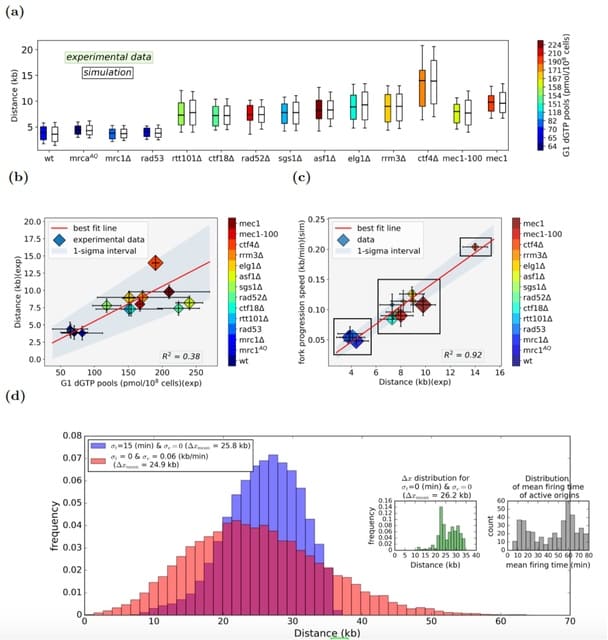
□ RepliSim: Computer simulations reveal mechanisms of spatio-temporal regulation of DNA replication
>> https://www.biorxiv.org/content/10.1101/2024.05.24.595841v1
RepliSim, a probabilistic numerical model for DNA replication simulation (RepliSim), which examines replication in the HU induced wt as well as checkpoint deficient cells.
The RepliSim model includes defined origin position, probabilistic initiation time and fork elongation rates assigned to origins and forks using a MonteCarlo method, and a transition time during the S-phase at which origins transit to a silent/non-active mode from being active.
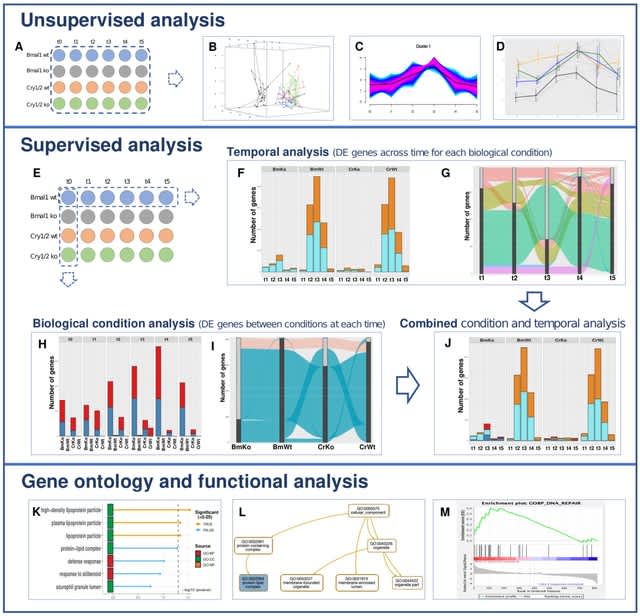
□ MultiRNAflow: integrated analysis of temporal RNA-seq data with multiple biological conditions
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae315/7684952
The MultiRNAflow suite gathers in a unified framework methodological tools found in various existing packages allowing to perform: i) exploratory (unsupervised) analysis of the data,
ii) supervised statistical analysis of dynamic transcriptional expression (DE genes), based on DESeq2 package and iii) functional and GO analyses of genes with gProfiler2 and generation of files for further analyses with several software.

□ Bayes factor for linear mixed model in genetic association studies
>> https://www.biorxiv.org/content/10.1101/2024.05.28.596229v1
IDUL (iterative dispersion update to fit linear mixed model) is designed for multi-omics analysis where each SNPs are tested for association with many phenotypes. IDUL has both theoretical and practical advantages over the Newton-Raphson method.
They transformed the standard linear mixed model as Bayesian linear regression, substituting the random effect by fixed effects with eigenvectors as covariates whose prior effect sizes are proportional to their corresponding eigenvalues.
Using conjugate normal inverse gamma priors on regression pa-rameters, Bayes factors can be computed in a closed form. The transformed Bayesian linear regression produced identical estimates to those of the best linear unbiased prediction (BLUP).
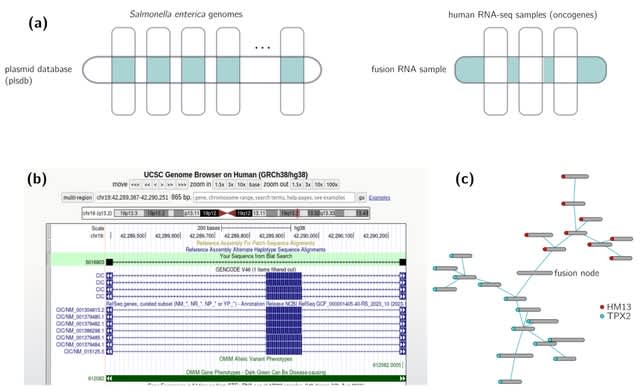
□ Constrained enumeration of k-mers from a collection of references with metadata
>> https://www.biorxiv.org/content/10.1101/2024.05.26.595967v1
A framework for efficiently enumerating all k-mers within a collection of references that satisfy constraints related to their metadata tags.
This method involves simplifying the query beforehand to reduce computation delays; the construction of the solution itself is carried out using CBL, a recent data structure specifically dedicated to the optimised computation of set operations on k-mer sets.
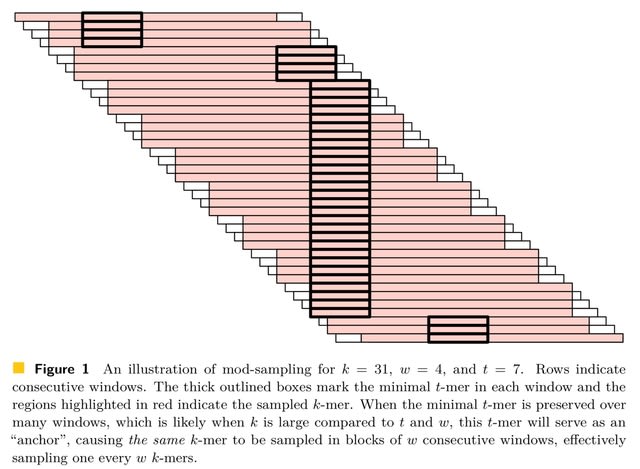
□ The mod-minimizer: a simple and efficient sampling algorithm for long k-mers
>> https://www.biorxiv.org/content/10.1101/2024.05.25.595898v1
mod-sampling, a novel approach to derive minimizer schemes. These schemes not only demonstrate provably lower density compared to classic random minimizers and other existing schemes but are also fast to compute, do not require any auxiliary space, and are easy to analyze.
Notably, a specific instantiation of the framework gives a scheme, the mod-minimizer, that achieves optimal density when k → ∞. The mod-minimizer has lower density than the method by Marçais et al. for practical values of k and w and converges to 1/w faster.
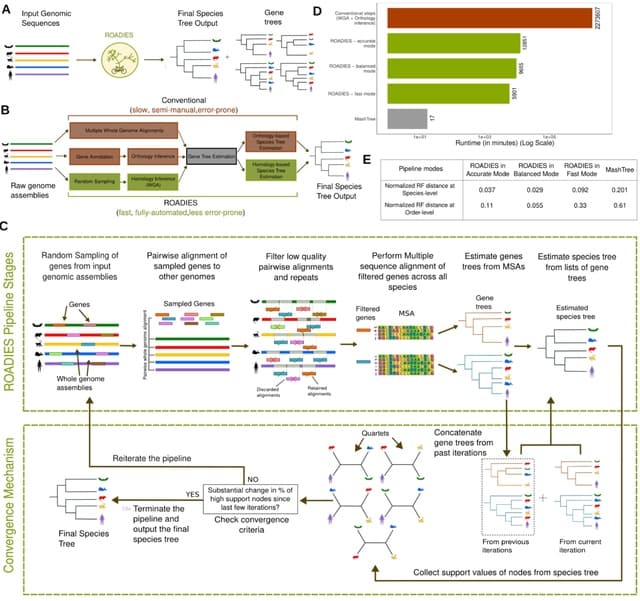
□ ROADIES: Accurate, scalable, and fully automated inference of species trees from raw genome assemblies
>> https://www.biorxiv.org/content/10.1101/2024.05.27.596098v1
ROADIES (Reference-free, Orthology-free, Alignment-free, Discordance-aware Estimation of Species Trees), a novel pipeline for species tree inference from raw genome assemblies that is fully automated, and provides flexibility to adjust the tradeoff between accuracy and runtime.
ROADIES eliminates the need to align whole genomes, choose a single reference species, or pre-select loci such as functional genes found using cumbersome annotation steps. ROADIES allows multi-copy genes, eliminating the need to detect orthology.
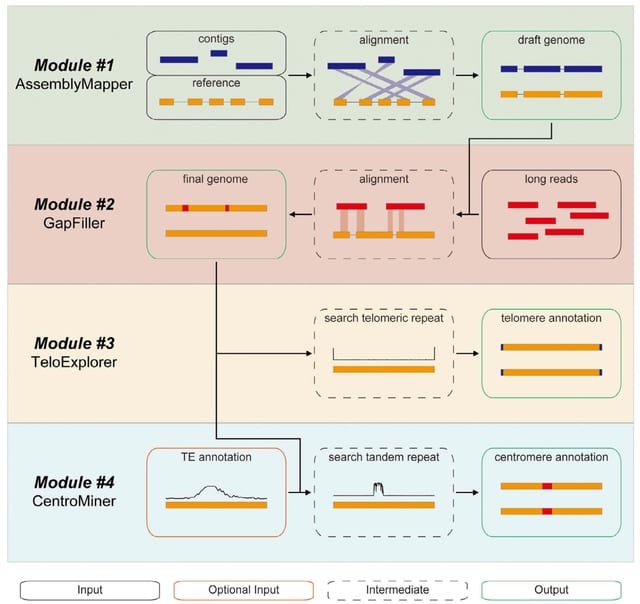
□ quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification
>> https://academic.oup.com/hr/article/10/8/uhad127/7197191
quarTeT, a user-friendly web toolkit specially designed for T2T genome assembly and characterization, including reference-guided genome assembly, ultra-long sequence-based gap filling, telomere identification, and de novo centromere prediction.
The quarTeT is named by the abbreviation 'Telomere-To-Telomere Toolkit' (TTTT), representing the combination of four modules: AssemblyMapper, GapFiller, TeloExplorer, and CentroMiner.
First, AssemblyMapper is designed to assemble phased cont chromosome-level genome by referring to a closely related genome.
Then, GapFiller would endeavor to fill all unclose given genome with the aid of additional ultra-long sequences. Finally, TeloExplorer and CentroMiner are applied to identif telomere and centromere as well as their localizations on each chromosome.

□ FinaleToolkit: Accelerating Cell-Free DNA Fragmentation Analysis with a High-Speed Computational Toolkit
>> https://www.biorxiv.org/content/10.1101/2024.05.29.596414v1
FinaleToolkit (FragmentatIoN AnaLysis of cEll-free DNA Toolkit) is a package and standalone program to extract fragmentation features of cell-free DNA from paired-end sequencing data.
FinaleToolkit can generate genome-wide WPS features from a ~100X cfDNA whole-genome sequencing (WGS) dataset in 1.2 hours using 16 CPU cores, offering up to a ~50-fold increase in processing speed compared to original implementations in the same dataset.

□ A Novel Approach for Accurate Sequence Assembly Using de Bruijn graphs
>> https://www.biorxiv.org/content/10.1101/2024.05.29.596541v1
Leveraging weighted de Bruin graphs as graphical probability models representing the relative abundances and qualities of kmers within FASTQ-encoded observations.
Utilizing these weighted de Bruijn graphs to identify alternate, higher-likelihood candidate sequences compared to the original observations, which are known to contain errors.
By improving the original observations with these resampled paths, iteratively across increasing k-lengths, we can use this expectation-maximization approach to "polish" read sets from any sequencing technology according to the mutual information shared in the reads.
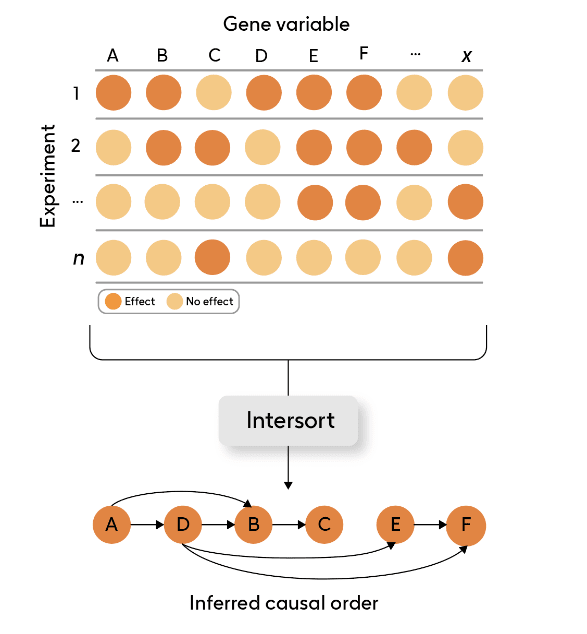
□ Intersort: Deriving Causal Order from Single-Variable Interventions: Guarantees & Algorithm
>> https://arxiv.org/abs/2405.18314
Intersort infers the causal order from datasets containing large numbers of single-variable interventions. Intersort relies on ε-interventional faithfulness, which characterizes the strength of changes in marginal distributions between observational and interventional distributions.
INTERSORT performs well on all data domains, and shows decreasing error as more interventions are available, exhibiting the model's capability to capitalize on the interventional information to recover the causal order across diverse settings.
ε-interventional faithfulness is fulfilled by a diverse set of data types, and that this property can be robustly exploited to recover causal information.
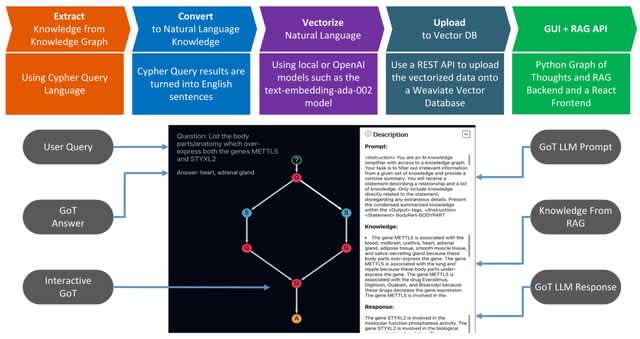
□ KRAGEN: a knowledge Graph-Enhanced RAG framework for biomedical problem solving using large language models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae353/7687047
KRAGEN (Knowledge Retrieval Augmented Generation ENgine) is a new tool that combines knowledge graphs, Retrieval Augmented Generation (RAG). KRAGEN uses advanced prompting techniques: namely graph-of-thoughts, to dynamically break down a complex problem into smaller subproblems.
KRAGEN embeds the knowledge graph information into vector embeddings to create a searchable vector database. This database serves as the backbone for the RAG system, which retrieves relevant information to support the generation of responses by a language model.
□ PanTools: Exploring intra- and intergenomic variation in haplotype-resolved pangenomes
>> https://www.biorxiv.org/content/10.1101/2024.06.05.597558v1
PanTools stores a distinctive hierarchical graph structure in a Neo4j database, including a compacted De Bruijn graph (DBG) to represent sequences. Structural annotation nodes are linked to their respective start and stop positions in the DBG.
The heterogeneous graph can be queried through Neo4j's Cypher query language. PanTools has a hierarchical pangenome representation, linking divergent genomes not only through a sequence variation graph but also through structural and functional annotations.

□ CellFM: a large-scale foundation model pre-trained on transcriptomics of 100 million human cells
>> https://www.biorxiv.org/content/10.1101/2024.06.04.597369v1
CellFM, a robust single-cell foundation model with an impressive 800 million param-eters, marking an eightfold increase over the current largest single-species model. CellFM is integrated with ERetNet, a Transformer architecture variant with linear complexity.
ERetNet Layers, each equipped with multi-head attention mechanisms that concurrently learn gene embeddings and the complex interplay between genes. CellFM begins by converting scalar gene expression data into rich, high-dimensional embedding features through its embedding module.

□ Systematic assessment of long-read RNA-seq methods for transcript identification and quantification
>> https://www.nature.com/articles/s41592-024-02298-3
ONT sequencing of CDNA and Cap Trap libraries produced many reads, whereas CDNA-PacBio and R2C2-ONT gave the most accurate ones.
For simulation data, tools performed markedly better on PacBio data than ONT data. FLAIR, IsoQuant, Iso Tools and TALON on cDNA-PacBio exhibited the highest correlation between estimation and ground truth, slightly surpassing RSEM and outperforming other long-read pipelines.
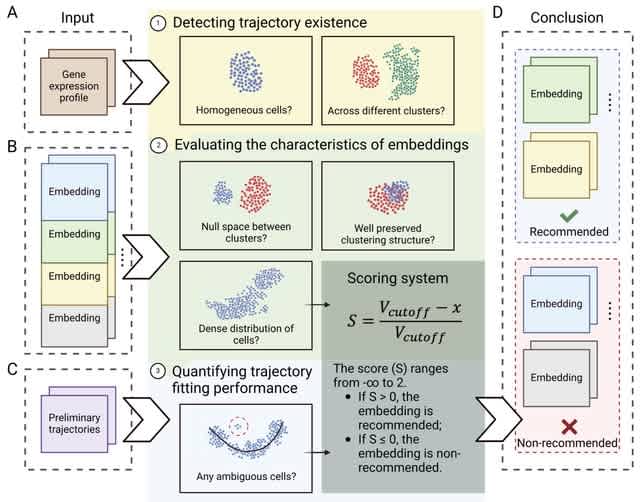
□ Escort: Data-driven selection of analysis decisions in single-cell RNA-seq trajectory inference
>> https://academic.oup.com/bib/article/25/3/bbae216/7667559
Escort is a framework for evaluating a single-cell RNA-seq dataset’s suitability for trajectory inference and for quantifying trajectory properties influenced by analysis decisions.
Escort detects the presence of a trajectory signal in the dataset before proceeding to evaluations of embeddings. In the final step, the preferred trajectory inference method of the user is used to fit a preliminary trajectory to evaluate method-specific hyperparameters.
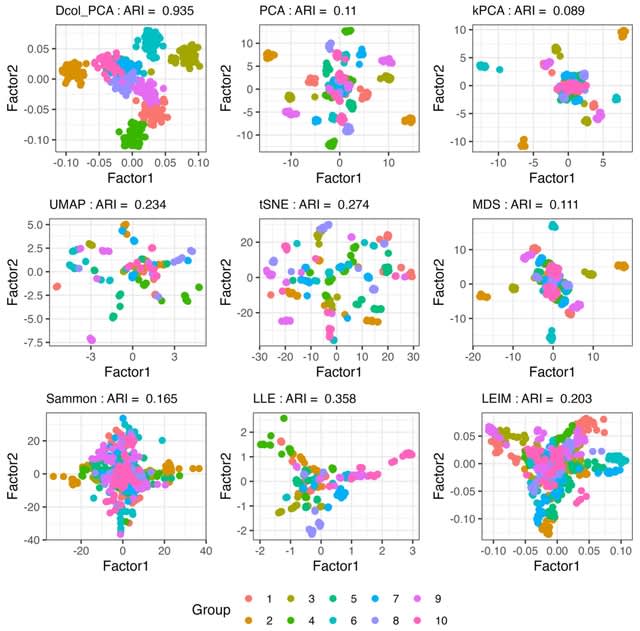
□ DCOL: Fast and Tuning-free Nonlinear Data Embedding and Integration
>> https://www.biorxiv.org/content/10.1101/2024.06.06.597744v1
DCOL (Dissimilarity based on Conditional Ordered List) correlation, a general association measure designed to quantify functional relationships between two random variables.
When two random variables are linearly related, their DCOL correlation essentially equals their absolute correlation value.
When the two random variables have other dependencies that cannot be captured by correlation alone, but one variable can be expressed as a continuous function of the other variable, DCOL correlation can still detect such nonlinear signals.

□ CelFiE-ISH: a probabilistic model for multi-cell type deconvolution from single-molecule DNA methylation haplotypes
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03275-x
CelFiE-ISH, which extends an existing method (CelFiE) to use within-read haplotype information. CelFiE-ISH jointly re-estimates the reference atlas along with the input samples ("ReAtlas" mode), similar to the default algorithm of CelFiE.
CelFiE-ISH had a significant advantage over CelFiE, as well as UXM, but only about 30% improvement, not nearly as strong as seen in the 2-state simulation model. But CelFiE-ISH can detect a cell type present in just 0.03% of reads out of a total of 5x genomic sequencing coverage.

□ quipcell: Fine-scale cellular deconvolution via generalized maximum entropy on canonical correlation features
>> https://www.biorxiv.org/content/10.1101/2024.06.07.598010v1
quipcell, a novel method for bulk deconvolution, that is a convex optimization problem and a Generalized Cross Entropy method. Quipcell represents each sample as a probability distribution over some reference single-cell dataset.
A key aspect of this density estimation procedure is the embedding space used to represent the single cells. Quipcell requires this embedding to be a linear transformation of the original single cell data.
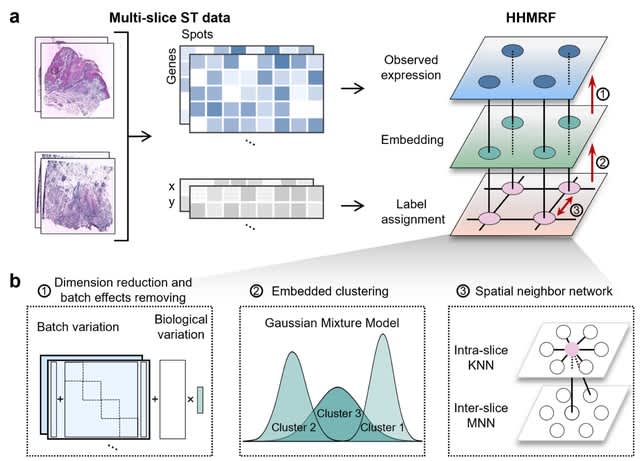
□ STADIA: Statistical batch-aware embedded integration, dimension reduction and alignment for spatial transcriptomics
>> https://www.biorxiv.org/content/10.1101/2024.06.10.598190v1
STADIA (ST Analysis tool for multi-slice integration, Dimension reduction and Alignment) is a hierarchical hidden Markov random field model (HHMRF) consisting of two hidden states: low-dimensional batch-corrected embeddings and spatially-aware cluster assignments.
STADIA first performs both linear dimension reduction and batch effect correction using a Bayesian factor regression model with L/S adjustment. Then, STADIA uses the GMM for embedded clustering.
STADIA applies the Potts model on an undirected graph, where nodes are spots from all slices and edges are intra-batch KNN pairs using coordinates and inter-batch MNN pairs using gene expression profiles.










※コメント投稿者のブログIDはブログ作成者のみに通知されます