









その肌は波立つことのない漆黒の海のようだった。見果てぬ弧で夜を2つに分ち、月の暈を纏いながらいよいよ彼の岸に泳ぎ渡らんとする者を拒んでいる。この水底は劫火よりも微温い熱を持ち、かつて亡骸であった星が、無数の裂け目に音も無く律動している。樹々のように目を塞ぎ、風のように声を遮った。

□ Poincare Maps for Analyzing Complex Hierarchies in Single-Cell Data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/02/689547.full.pdf
Poincaré maps, a method harnessing the power of hyperbolic space into the realm of single-cell data analysis.
Often understood as a continuous extension of trees, hyperbolic geometry enables the embedding of complex hierarchical data in as few as two dimensions and well-preserves distances between points in the hierarchy.
This enables direct exploratory analysis and the use of our embeddings in a wide variety of downstream data analysis tasks, such as visualization, clustering, lineage detection and pseudotime inference.

□ A novel metric reveals previously unrecognized distortion in dimensionality reduction of scRNA-Seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/02/689851.full.pdf
a straightforward approach to quantifying this distortion by comparing the local neighborhoods of points before and after dimensionality reduction.
The first applied approach to the simple problem of embedding points on the surface of a hypersphere into the appropriate latent dimension from a higher-dimensional space.
trivially embed those points into a 100-dimensional space by just adding 80 zeroes to the end of those vectors.

□ circDeep: Deep learning approach for circular RNA classification from other long non-coding RNA
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz537/5527751
an End-to-End deep learning framework, circDeep, to classify circular RNA from other lncRNA.
circDeep fuses an RCM descriptor, ACNN-BLSTM sequence descriptor, and a conservation descriptor into high level abstraction descriptors, where the shared representations across different modalities are integrated.
□ Raptor: Graph-based mapping of long sequences, noisy or HiFi
>> https://github.com/isovic/raptor
Raptor is a very versatile and fast graph based sequence mapper/aligner with a large number of features, Sequence-to-Graph mapping and path alignment.
Raptor can currently read both Graphical Fragment Assembly (GFA)-1 and GFA-2 formats to define the graph. Graph-based dynamic programming is applied on the AnchorGraph to chain the anchors over the graph.
□ Mapping Vector Field of Single Cells
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/09/696724.full.pdf
a new framework that combines promoter state fluctuations, RNA transcription, metabolic labeling, splicing, translation, and RNA/protein degradation to infer expression dynamics at scale.
and can reconstruct functional vector fields in the high-dimensional state space from sparse vector samples.
This vector field reconstruction method also directly enables global mapping of potential landscapes that reflects the relative stability of a given cell state, and the minimal transition time and most probable paths between any cell states in the state space.

□ Topological quantum matter in synthetic dimensions
>> https://www.nature.com/articles/s42254-019-0045-3
This approach provides a way to engineer lattice Hamiltonians and enables the realization of higher-dimensional topological models in platforms with lower dimensionality.
The main idea of a synthetic dimension is to couple together suitable degrees of freedom, such as a set of internal atomic states, in order to mimic the motion of a particle along an extra spatial dimension.
□ An Introduction to Higher Categorical Algebra
>> https://arxiv.org/pdf/1907.02904v1.pdf
symmetric monoidal stable ∞-categories, such as the derived ∞-category of a commutative ring, before turning to the main example, the ∞-category of spectra.
the functors which comprise the cohomology theory are represented by the spaces of the infinite delooping.
□ Minimal time sliding mode control for evolution equations in Hilbert spaces
>> https://arxiv.org/pdf/1906.11918v1.pdf
by the time optimal control problem we mean to search for a constrained internal controller able to drive the trajectory of the solution from an initial state to a given target set in the shortest time, while controlling over the complete timespan.
In time optimal control the optimality criterion is the elapsed time. A time optimal control for a family of evolution equations in Hilbert spaces.
The existence of the optimal time control for a phase-field system for a regular double-well potential, by using the Carleman inequality and the maximum principle was established by using two controls acting in subsets of the space domain.


□ Random Surfaces Hide an Intricate Order
>> https://www.quantamagazine.org/random-surfaces-hide-an-intricate-order-20190702
Because the underlying surface is chosen at random, and the process of coloring the vertices is random, the largest cluster on one surface will always be different from the largest cluster on another.
across all surfaces and all possible ways of coloring the vertices on those surfaces, the largest clusters have traits in common.

□ A computational framework for a Lyapunov-enabled analysis of biochemical reaction networks
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/09/696716.full.pdf
a class of networks that are “structurally (mono) attractive” by which we mean that they are incapable of exhibiting multiple steady states, oscillation, or chaos by the virtue of their reaction graphs.
These networks are characterized by the existence of a universal energy-like function which we call a Robust Lyapunov function (RLF).
Lyapunov-Enabled Analysis of Reaction Networks (LEARN), is provided that constructs such functions or rules out their existence.
□ Fused Sparse SEM: Inference of Differential Gene Regulatory Networks Based on Gene Expression and Genetic Perturbation Data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz529/5526871
Gene regulatory networks (GRNs) with the structural equation model that can integrate gene expression and genetic perturbation data, and develop an algorithm - fused sparse SEM (FSSEM), to jointly infer GRNs under two conditions, and then to identify difference of the two GRNs.
When the objective function in an optimization problem is non-convex and non-smooth, it is possible that the coordinate descent method fails to converge.
the FSSEM algorithm converges to a stationary point, because the objective function satisfies the conditions for the convergence of the PALM method.
□ Empirical Performance of Tree-based Inference of Phylogenetic Networks
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/05/693986.full.pdf
combining the strengths of the two—the speed of tree-based inference and the accuracy of the divide- and-conquer approach—could provide a promising approach to large-scale network inference.
the start tree built from inferred gene trees using ASTRAL- III is much better than concatenation using IQ-TREE. This is because the rate of ILS is high, and ASTRAL-III considers the gene tree topology conflicts.
□ scVILP: A Combinatorial Approach for Single-cell Variant Detection via Phylogenetic Inference
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/05/693960.full.pdf
scVILP (single-cell Variant calling via Integer Linear Program) assumes that the somatic cells evolve along a phylogenetic tree and mutations are acquired along the branches following the infinite sites model as have been used in previous bulk and single-cell studies.
The supertree-based approach is deterministic and solve the problem using a novel Integer Linear Program (ILP) that achieves similar accuracy as SCIΦ but performs significantly better than SCIΦ in terms of runtime.
identify the set of single-nucleotide variants in the single cells and genotype them in such a way so that it maximizes the probability of the observed read counts and also the cells are placed at the leaves of a perfect phylogeny that satisfies the Infinite Sites Assumption (ISA).

□ DolphinNext: A distributed data processing platform for high throughput genomics
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/02/689539.full.pdf
The guiding principle of DolphinNext is to facilitate the building and deployment of complex pipelines using a modular approach implemented in a graphical interface.
DolphinNext provides seamless portability to distributed computational environments such as high performance clusters or cloud computing environments.


□ Characterizing RNA stability genome-wide through combined analysis of PRO-seq and RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/02/690644.full.pdf
RNA splicing-related features, including intron length, are positively correlated with RNA stability, whereas features related to miRNA binding, DNA methylation, and G+C-richness are negatively correlated with RNA stability.
a measure of predicted stability based on U1 binding sites and polyadenylation sites distinguishes between unstable noncoding and stable coding transcripts but is not predictive of relative stability within the mRNA or lincRNA classes.

□ uap: Reproducible and Robust HTS Data Analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/02/690438.full.pdf
uap (Universal Analysis Pipeline) is the workflow management system that may be 59 used to implement any DAG-like data analysis workflow, but is primarily aimed at HTS data analysis.
provide a uap configuration file for combining split-read mapping with de novo transcript assembly.
uap reads the sequencing data either from an Illumina sequencing run folder, or a set of fastq files, applies quality control, removes adapter sequences, and maps the reads to a genome using tophat2 and segemehl.

□ SRAssembler: Selective Recursive local Assembly of homologous genomic regions
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2949-4
The workflow implements a recursive strategy by which relevant reads are successively pulled from the input sets based on overlapping significant matches, resulting in virtual chromosome walking.
The program can also aid decision making on the depth of sequencing in an ongoing novel genome sequencing project or with respect to ultimate whole genome assembly strategies.

□ DNA assembly for nanopore data storage readout
>> https://www.nature.com/articles/s41467-019-10978-4
an approach for decoding information stored in DNA that combines random-access, DNA assembly and nanopore sequencing.
This Gibson Assembly concatenation strategy is generalizable to any short amplicon sequencing application where higher nanopore sequencing throughput is desirable.
Read until decoding of 1.67 megabytes of information stored in short fragments of synthetic DNA using a portable nanopore sequencing platform.

□ Automated methods enable direct computation on phenotypic descriptions for novel candidate gene prediction
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/03/689976.full.pdf
These representations include the EQ (Entity-Quality) formalism, which uses terms from biological ontologies to represent phenotypes in a standardized, semantically-rich format, as well as numerical vector representations generated using Natural Language Processing (NLP) methods.
Computationally derived EQ and vector representations were comparably successful in recapitulating biological truth to representations created through manual EQ statement curation.

□ Dynamics and Topology of Human Transcribed Cis-regulatory Elements
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/03/689968.full.pdf
a simple and robust approach to globally determine 5’-ends of nascent RNAs (NET-CAGE) in diverse cells and tissues, thereby sensitively detecting unstable transcripts including enhancer-derived RNAs.
By integrating NET-CAGE data with chromatin interaction maps, cis-regulatory elements are topologically connected according to their cell-type specificity.

□ tappAS: A comprehensive computational framework for the analysis of the functional impact of differential splicing
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/03/690743.full.pdf
a novel computational framework for the study AltTP from a functional perspective, introducing the Functional Iso-Transcriptomics (FIT) analysis approach.
This framework uses a rich isoform-level annotation database of functional domains, motifs and sites –both coding and non- coding- and introduces novel analysis methods to interrogate different aspects of the functional relevance of isoform complexity.

□ Genetic Variation, Comparative Genomics, and the Diagnosis of Disease
>> https://www.nejm.org/doi/full/10.1056/NEJMra1809315
The discovery of pathogenic variation and its mechanism of action often is less trivial, and decades of research can be required in order to identify the variants underlying both mendelian and complex genetic traits.
There are three key aspects to genetic disease associations: comprehensive variant discovery, accurate allele-frequency determination, and an understanding of the pattern of normal variation and its effect on expression.

□ Locating the source node of diffusion process in cyber-physical networks via minimum observers
>> https://aip.scitation.org/doi/10.1063/1.5092772
a greedy optimization algorithm by analyzing the difference of propagation delay between each pair of observers.
Combining this greedy algorithm with the diffusion-back method provides a framework that outperforms other strategies for locating the source node in cyber physical networks.

□ Deep Learning For Denoising Hi-C Chromosomal Contact Data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/04/692558.full.pdf
unsupervised and semi-supervised deep learning algorithms (i.e. deep convolutional autoencoders) to denoise Hi-C contact matrix data and improve the quality of chromosome structure predictions.
the network considered is a denoising autoencoder, a flavor of unsupervised learning, rather than the supervised deep network used in HiCNN and HiCPlus.
□ MethylNet: A Modular Deep Learning Approach to Methylation Prediction
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/04/692665.full.pdf
MethylNet is a deep learning latent space regression and classification tasks through the development of a modular framework.
MethylNet framework enables rapid production-scale research and development in the deep learning epigenetic space.
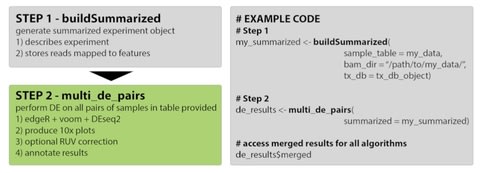
□ consensusDE: an R package for assessing consensus of multiple RNA-seq algorithms with RUV correction
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/04/692582.full.pdf
Removal of unwanted variation (RUV) has also been proposed as a method for stabilizing differential expression (DE) results.
consensusDE integrates DE results from edgeR, limma/voom and DEseq2 easily and reproducibly, with the additional option of integrating RUV.
□ BioGD: Bio-inspired robust gradient descent
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0219004
BioGD inspired by the stability and adaptability of biological systems to unknown and changing environments.
The proposed optimization technique involves an open-ended adaptation process with regard to two hyperparameters inherited from the generalized Verhulst population growth equation.
□ Minigraph: Proof-of-concept seq-to-graph mapper and graph generator
>> https://github.com/lh3/minigraph
Minigraph finds approximate locations of a query sequence in a sequence graph and incrementally augments an existing graph with long query subsequences diverged from the graph.
The minigraph Graphical Fragment Assembly (GFA) parser seamlessly parses FASTA and converts it to GFA internally, and also provide sequences in FASTA as the reference. In this case, minigraph will behave like minimap2 but without base-level alignment.
□ New contributions to the Hamiltonian and Lagrangian contact formalisms for dissipative mechanical systems and their symmetries
>> https://arxiv.org/pdf/1907.02947.pdf
a geometric framework for the Lagrangian formalism of dissipative autonomous mechanical systems using contact geometry.
a new form of the contact Hamiltonian and Lagrangian equations, and compare the two Lagrangian formalisms existing in the literature, proving their equivalence.


□ Synthetic Genetic Codes Designed to Hinder Evolution
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/08/695569.full.pdf
a set of “fail-safe” genetic codes designed to map mutations to deleterious phenotypes, independent of the biological system in which these codes are implemented.
fail-safe codes supporting expression of 20 or 15 amino acids could slow the evolution of proteins in so-encoded organisms to 30% or 0% the rate of standard-code organisms.

□ Bifrost – Highly parallel construction and indexing of colored and compacted de Bruijn graphs
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/08/695338.full.pdf
Bifrost features a broad range of functions such as sequence querying, storage of user data alongside vertices and graph editing that automatically preserve the compaction property.
Bifrost is about eight times faster than VARI-merge and uses about 20 times less memory with no external disk.
Bifrost is competitive with the state-of- the-art de Bruijn graph construction method BCALM2 and the unitig indexing tool Blight with the advantage that Bifrost is dynamic.

□ PheGWAS: A new dimension to visualize GWAS across multiple phenotypes
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/08/694794.full.pdf
PheGWAS was developed to enhance exploration of phenome-wide pleiotropy at the genome-wide level through the efficient generation of a dynamic visualization combining Manhattan plots from GWAS with PheWAS to create a three-dimensional “landscape”.
Pleiotropy in sub-surface GWAS significance strata can be explored in a sectional view plotted within user defined levels.
□ Deconvolution of autoencoders to learn biological regulatory modules from single cell mRNA sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2952-9
The model can, from scRNA-seq data, delineate biological meaningful modules that govern a dataset, as well as give information as to which modules are active in each single cell.
In comparison with other dimensionality reduction methods, this approach has the benefit of both handling well the zero-inflated nature of scRNA-seq, and validating that the model captures relevant information, by establishing a link between input and decoded data.
□ STARRPeaker: Uniform processing and accurate identification of whole human STARR-seq active regions
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/08/694869.full.pdf
a statistical framework for uniformly processing STARR-seq data: STARRPeaker, outperforms other peak callers in terms of identifying known enhancers.
STARRPeaker statistically models the basal level of transcription, accounting for potential confounding factors, and accurately identifies reproducible enhancers.
□ SMURF-seq: efficient copy number profiling on long-read sequencers
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1732-1
SMURF-seq, a protocol to efficiently sequence short DNA molecules on a long-read sequencer by randomly ligating them to form long molecules.
Applying SMURF-seq using the Oxford Nanopore MinION yields up to 30 fragments per read, providing an average of 6.2 and up to 7.5 million mappable fragments per run, increasing information throughput for read-counting applications.
□ Transcriptome assembly from long-read RNA-seq alignments with StringTie2
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/08/694554.full.pdf
StringTie2 also offers the ability to work with full-length super-reads assembled from short reads, which further improves the quality of assemblies.
StringTie2 on average correctly assembles 8.3 and 2.6 times as many transcripts as FLAIR and Traphlor, respectively, with substantially higher precision.
□ ConsHMM: Systematic discovery of conservation states for single-nucleotide annotation of the human genome
>> https://www.nature.com/articles/s42003-019-0488-1
ConsHMM applies a multivariate hidden Markov model to learn de novo ‘conservation states’ based on the combinatorial and spatial patterns of which species align to and match a reference genome in a multiple species DNA sequence alignment.
ConsHMM assumes that the probability of observing a specific combination of observations is determined by a product of independent multinomial random variables.

□ Enter the Matrix: Factorization Uncovers Knowledge from Omics
>> https://www.cell.com/trends/genetics/fulltext/S0168-9525(18)30124-0
MF is also referred to as matrix decomposition, and the corresponding inference problem as deconvolution.
MFs learn two sets of low-dimensional representations (in each matrix factor) from high-dimensional data: one defining molecular relationships (amplitude) and another defining sample-level relationships (pattern).
Clustering, subtype discovery, in silico microdissection, and timecourse analysis are all enabled by analysis of the pattern matrix.
□ Detecting Transcriptomic Structural Variants in Heterogeneous Contexts via the Multiple Compatible Arrangements Problem
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/09/697367.full.pdf
MULTIPLE COMPATIBLE ARRANGEMENT PROBLEM (MCAP) seeks a given k, an optimal set of k arrangements of segments from GSG such that number of consistent read alignments is maximized, where each arrangement describes the permutation of all segments and orientation of each segment.
an integer linear programming formulation for general k.
MCAP is NP-hard and provide an 1/4-approximation algorithm for k=1 and a 3/4-approximation algorithm for the diploid case (k=2) assuming an oracle for k=1.

□ ReCappable Seq: Comprehensive Determination of Transcription Start Sites derived from all RNA polymerases
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/09/696559.full.pdf
ReCappable-seq reveals distinct epigenetic marks among Pol-lI and non-Pol-II TSS and provides a unique opportunity to concurrently interrogate the regulatory landscape of coding and non-coding RNA.
□ Look4TRs: A de-novo tool for detecting simple tandem repeats using self-supervised hidden Markov models
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz551/5530162
Look4TRs adapts itself to the input genomes, balancing high sensitivity and low false positive rate. It auto-calibrates itself.










※コメント投稿者のブログIDはブログ作成者のみに通知されます