









私たちが直面する問題は2種類に分けられる。それは「己の限界」と「他者の檻」である。全ての主観者が『反復』するプロセスを織り込んで、2つの問題は常に背中合わせとなる。自らが解決した問題は常に他者を囚え続け、鏡のようにその逆が成り立つ。檻から出た先は檻であり、入れ子のように循環する。
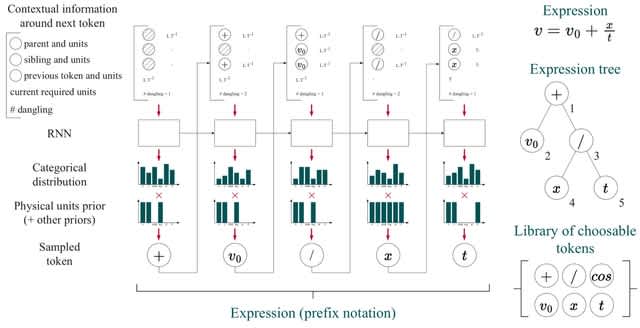
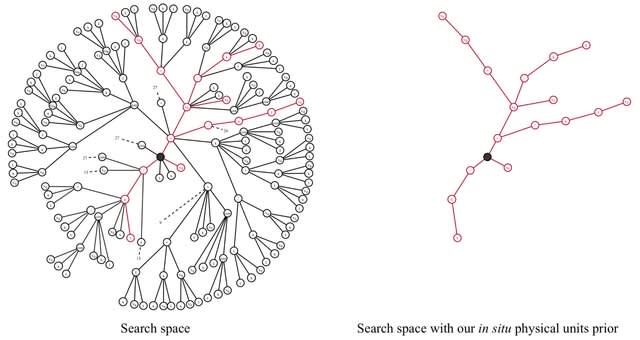

□ Φ-SO: Deep symbolic regression for physics guided by units constraints: toward the automated discovery of physical laws
>> https://arxiv.org/abs/2303.03192
Φ-SO, a Physical Symbolic Optimization framework for recovering analytical symbolic expressions from physics data using deep reinforcement learning techniques by learning units constraints.
Φ-SO restricts the freedom of the equation generator, and balanced units are proposed by construction, thus greatly reducing the search space. It enables the algorithm to zero-out the probability of forbidden symbols that would result in expressions that violate units rules.
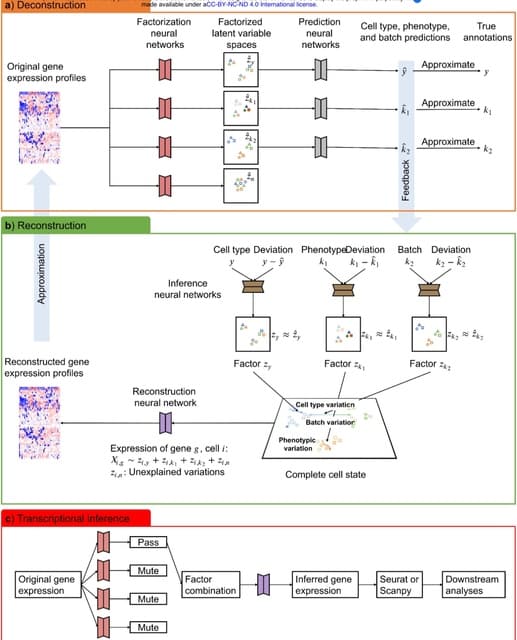
□ scPheno: Extraction of biological signals by factorization enables the reliable analysis of single-cell transcriptomics
>> https://www.biorxiv.org/content/10.1101/2023.03.04.531126v1
scPheno, a deep auto-regressive factor model that is used to extract the biological signals imbedded in transcriptome, identify gene expression variations associated with each of the phenotypes, and re-build the accumulative effect of multiple phenotypes on cell states.
scPheno will factorize gene expression pertaining to a phenotypic factor and project cells onto a latent variable space, where the latent variable specifies a hidden cell state and cells of the same hidden states will cluster together.
The deep factor model will infer the factorized latent variable spaces. The factorization neural networks and the reconstruction neural network can be coupled to predict gene expression in relation to any factor combination.

□ INSnet: a method for detecting insertions based on deep learning network
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05216-0
INSnet divides the reference genome into continuous sub-regions and takes five features for each locus through alignments between long reads and the reference genome. Next, INSnet uses a depthwise separable convolutional network.
INSnet uses two attention mechanisms, the convolutional block attention module (CBAM) and efficient channel attention (ECA) to extract key alignment features in each sub-region. INSnet uses a gated recurrent unit (GRU) network to further extract more important SV signatures.
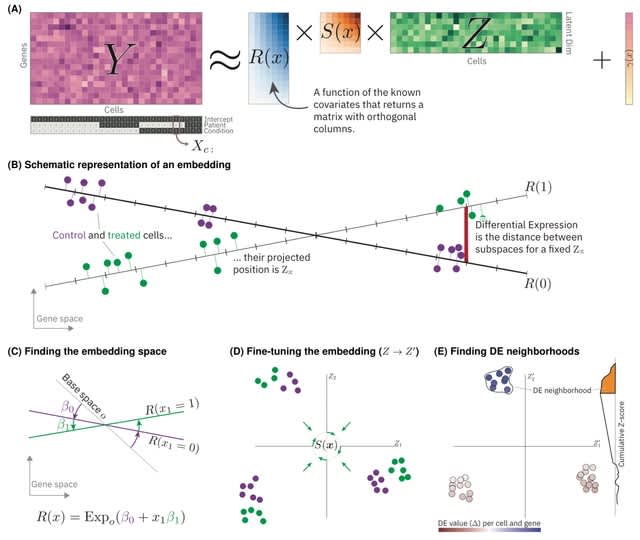
□ LEMUR: Analysis of multi-condition single-cell data with latent embedding multivariate regression
>> https://www.biorxiv.org/content/10.1101/2023.03.06.531268v1
A new statistical model for differential expression analysis (or ANOVA) of multi-condition single-cell data that combines the ideas of linear models and principal compo- nent analysis (PCA).
Latent embedding multivariate regression (LEMUR) is based on a parametric mapping of latent space representations into each other and uses a design matrix to encode categorical and continuous covariates.

□ The Network Zoo: a multilingual package for the inference and analysis of gene regulatory networks
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02877-1
The Network Zoo, a platform that harmonizes the codebase for these methods, in line with recent similar efforts, and provides implementations in R, Python, MATLAB, and C. The netZoo codebase has helped develop an ecosystem of online resources for GRN inference and analysis.
netZoo integrates PANDA, LIONESS, and MONSTER to infer TF-gene targeting to explore how regulatory changes affect disease phenotype, and used DRAGON to integrate nine types of genomic information and find multi-omic markers that are associated with drug sensitivity.
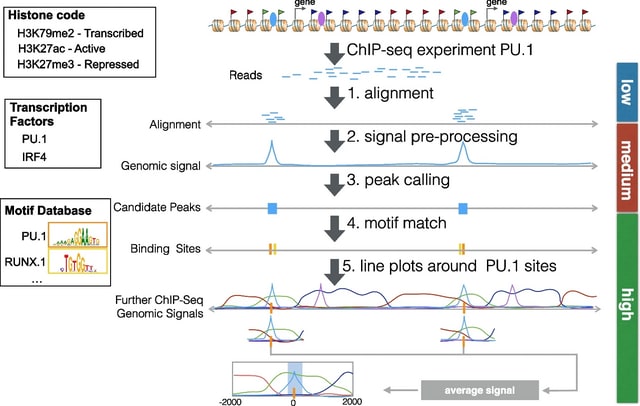
□ RGT: a toolbox for the integrative analysis of high throughput regulatory genomics data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05184-5
Regulatory Genomics Toolbox (RGT) was programmed in an oriented-object fashion and its core classes provided functionalities to handle typical regulatory genomics data: regions and signals.
RGT built distinct regulatory genomics tools, i.e., HINT for footprinting analysis, TDF for finding DNA–RNA triplex, THOR for ChIP-seq differential peak calling, motif analysis for TFBS matching and enrichment, and RGT-viz for regions association tests and data visualization.
THOR is a Hidden Markov Model-based approach to detect and analyze differential peaks in two sets of ChIP-seq data from distinct biological conditions with replicates. Triplex Domain Finder (TDF) characterizes the triplex-forming potential between RNA and DNA regions.
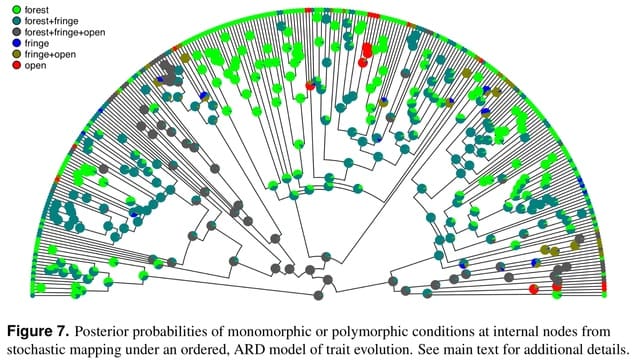
□ phytools 2.0: An updated R ecosystem for phylogenetic comparative methods (and other things)
>> https://www.biorxiv.org/content/10.1101/2023.03.08.531791v1
The phytools library has now grown to be very large – consisting of hundreds of functions, a documentation manual that’s over 200 pages in length, and tens of thousands of lines of computer code.
For Mk model-fitter (which here will be the phytools function fitMk), and for the other discrete character methods of the phytools R package, the input phenotypic trait data will typically takes the form of a character or factor vector.
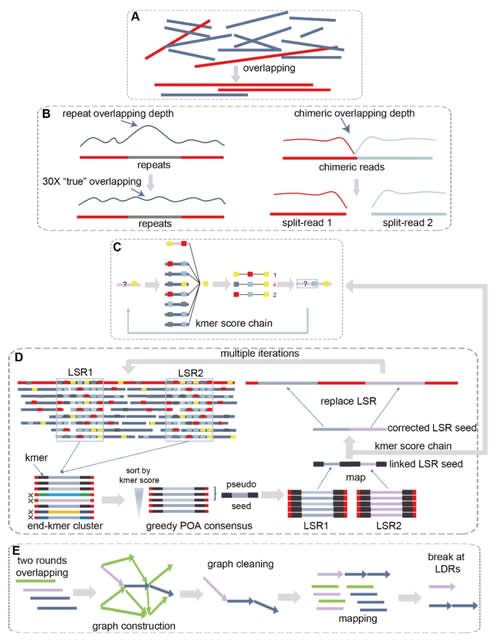
□ NextDenovo: An efficient error correction and accurate assembly tool for noisy long reads
>> https://www.biorxiv.org/content/10.1101/2023.03.09.531669v1
NextDenovo, a highly efficient error correction and CTA-based assembly tool for noisy long reads. NextDenovo can rapidly correct reads; these corrected reads contain fewer errors than other comparable tools and are characterized by fewer chimeric alignments.
NextDenovo uses the BOG algorithm to remove edges for non-repeat nodes. The graph usually contained some linear paths connecting some complex subgraphs. All paths were broken at the node connecting with multi-paths, and contigs were outputted from these broken linear paths.
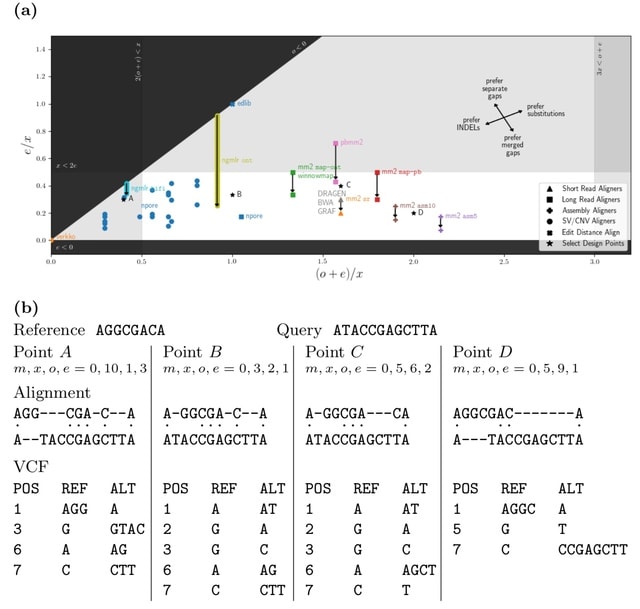
□ vcfdist: Accurately benchmarking phased small variant calls in human genomes
>> https://www.biorxiv.org/content/10.1101/2023.03.10.532078v1
vcfdist, an alignment-based small variant calling evaluator that standardizes query and truth VCF variants to a consistent representation, requires local phasing of both input VCFs, and gives partial credit to variant calls which are mostly (but not exactly) correct.
A novel variant clustering algorithm reduces downstream computation while discovering long range variant dependencies. A novel alignment distance based metrics which are independent of variant representation, and measure the distance b/n the final diploid truth / query sequences.
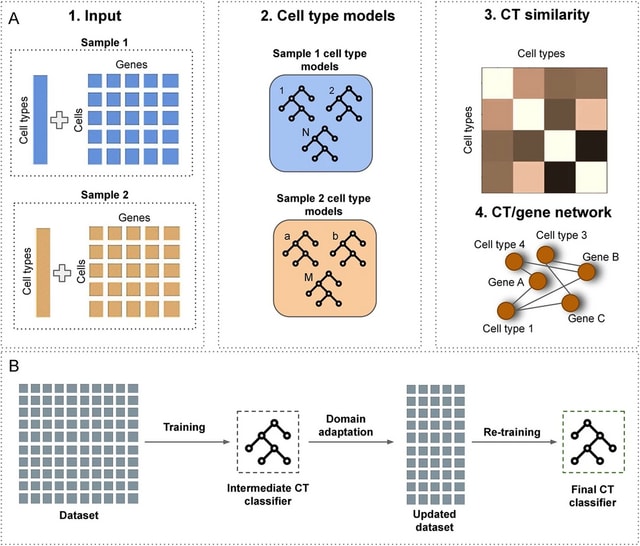
□ scEvoNet: a gradient boosting-based method for prediction of cell state evolution
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05213-3
scEvoNet, a method that builds a cell type-to-gene network using the Light Gradient Boosting Machine (LGBM) algorithm overcoming different domain effects (different species/different datasets) and dropouts that are inherent for the scRNA-seq data.
ScEvoNet builds the confusion matrix of cell states and a bipartite network connecting genes and cell states. It allows a user to obtain a set of genes shared by the characteristic signature of two cell states even between distantly-related datasets.
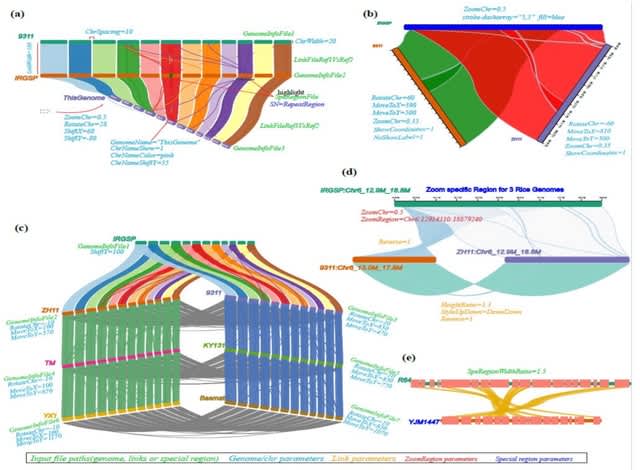
□ NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad121/7072460
NGenomeSyn, an easy-to-use and flexible tool, for publication-quality visualization of syntenic relationships (user-defined or generated by our custom script) and genomic features (e.g. repeats, structural variations, genes) on tens of genomes with high customization.
NGenomeSyn allows its user to adjust default options for genome and link styles defined in the configuration file and simply adjusts options of moving, scaling, and rotation of target genomes, yielding a rich layout and publication-ready figure.
□ containX: Coverage-preserving sparsification of overlap graphs for long-read assembly
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad124/7074174
ContainX heuristics are promising in terms of improving assembly quality by avoiding coverage gaps. The string graph model filters out contained reads during graph construction.
containX is a prototype implementation of an algorithm that decides which contained reads can be dropped during overlap graph sparsfication. Reads which are substrings of longer reads are typically referred to as contained reads.
Hifiasm retained fewer contained reads than ContainX but it failed to resolve a majority of coverage gaps. The unitig graph of Hifiasm has the least number of junction reads because it does additional graph pruning which is necessary for computing longer unitigs.
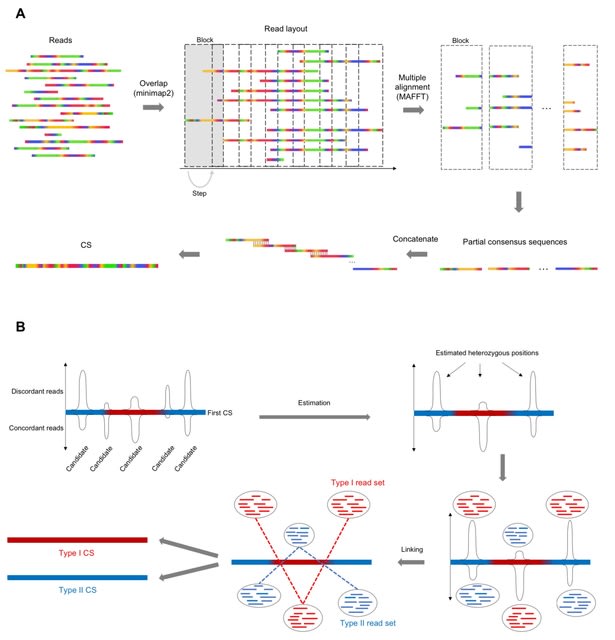
□ LoMA: Localized assembly for long reads enables genome-wide analysis of repetitive regions at single-base resolution in human genomes
>> https://pubmed.ncbi.nlm.nih.gov/36895025/
LoMA constructs a CS spanning a target region. This process is initiated by finding overlaps of raw reads using pairwise all-to-all alignment of minimap2, followed by a layout of overlapped reads. It divides the layout into multiple blocks to make partial consensus sequences.
LoMA captures haplotype structures based on SVs and produces haplotype-resolved CSs. LoMA predicts heterozygous loci in the region based on the extent of deviation from the binomial distribution, and the reads derived from each estimated haplotype are gathered.
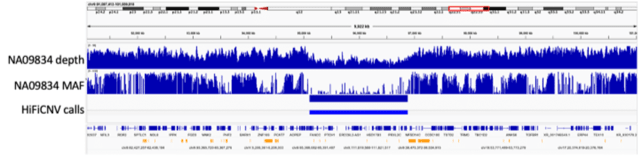
□ HiFiCNV : Copy number variant caller and depth visualization utility for PacBio HiFi reads
>> https://www.pacb.com/blog/hificnv/
HiFiCNV can generate several CNV related track files which can be loaded into IGV for visualization and assessment of its variant calls. HiFiCNV detected all large CNVs from this dataset, and 90% of those calls had high overlap accuracy when compared to the reported CNV.
Segmentation is performed by a Viterbi parse of the depth bins assuming the bin depth represents a Poisson sampling from a mean depth based on haploid depth. The haploid depth is computed from the zero-excluded mean depth of this chromosome set.

□ ReCo: automated NGS read-counting of single and combinatorial CRISPR gRNAs.
>> https://www.biorxiv.org/content/10.1101/2023.03.09.530923v1
ReCo! finds gRNA read counts (ReCo) in fastq files and runs as a standalone script or a python package. It can be used for single and combinatorial CRISPR-Cas libraries that have been sequenced with single-end or paired-end sequencing strategies.
ReCo works with conventionally cloned CRISPR-Cas libraries and 3Cs/3Cs-MPX libraries. ReCo can process multiple samples in a single run. It automatically determines the constant regions flanking the gRNAs, and utilizes Cutadapt to trim the fastq files.
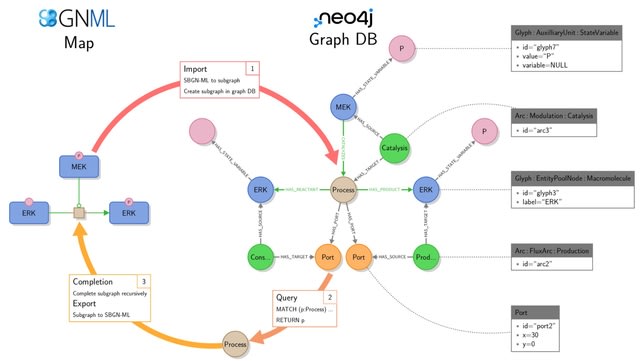
□ StonPy: a tool to parse and query collections of SBGN maps in a graph database
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad100/7075543
The StonPy library allows users to store SBGN-ML maps into a running Neo4j database, and conversely retrieve them into SBGN-ML. StonPy includes a completion module that allows users to build valid SBGN maps from query results representing parts of maps automatically.
SBGN arcs are optionally modelled using additional Neo4j relationships that mimic the structure of the SBGN map. StonPy brings new capabilities for storing and analyzing large collections of CellDesigner and SBGN maps using Neo4j and Cypher.
□ SLEMM: million-scale genomic predictions with window-based SNP weighting
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad127/7075542
SLEMM (Stochastic-Lanczos-Expedited Mixed Models) uses the Stochastic Lanczos REML and SNP effects for large datasets. SLEMM is fast enough for million-scale genomic predictions.
SLEMM with SNP weighting had overall the best predictive ability among a variety of genomic prediction methods including GCTA’s empirical BLUP, BayesR, KAML, and LDAK’s BOLT and BayesR models.

□ scDeepInsight: a supervised cell-type identification method for scRNA-seq data with deep learning
>> https://www.biorxiv.org/content/10.1101/2023.03.09.531861v1
scDeepInsight can directly annotate the query dataset based on the model trained on the reference dataset. scDeepInsight does preprocessing of scRNA-seq data, including quality control and integration through batch normalization.
scDeepInsight is a single-cell labeling model based on supervised learning, so a reference dataset is also required. DeepInsight is utilized to convert the processed non-image data into images.
□ A general minimal perfect hash function for canonical k-mers on arbitrary alphabets with an application to DNA sequences
>> https://www.biorxiv.org/content/10.1101/2023.03.09.531845v1
A minimal perfect hash function of canonical k-mers on alphabets of arbitrary size, i.e., a mapping to the interval [0, σk /2−1]. The approach is introduced for canonicalization under reversal and extended to canonicalization under reverse complementation.
The encoding is based on the observation that there are fewer canonical k-mers than there are k-mers in general. A mapping is only required if k-mer x is canonical, i.e., x is lexicographically smaller than or equal to x^−1.
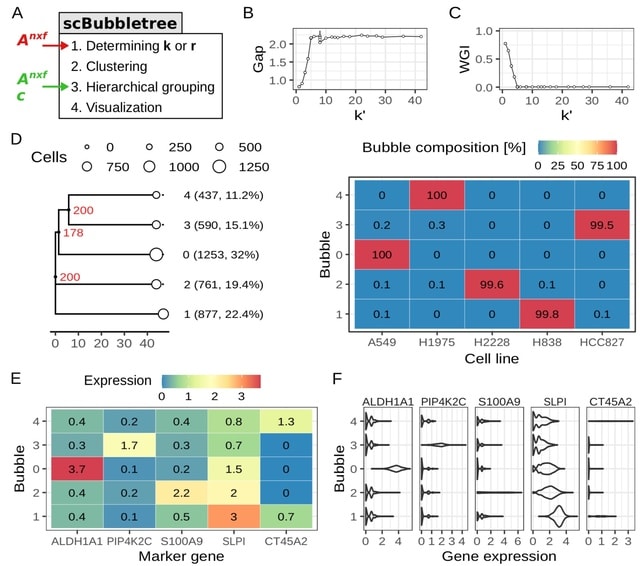
□ scBubbletree: quantitative visualization of single cell RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2023.03.09.531263v1
scBubbletree, a new scalable method for visualization of scRNA-seq data. The method identifies clusters of cells of similar transcriptomes and visualizes such clusters as “bubbles” at the tips of dendrograms, corresponding to quantitative summaries of cluster properties.
scBubbletree stacks bubble trees w/ further cluster-associated information. scBubbletree relies on the gap statistic method. scBubbletree can cluster scRNA-seq data in two ways, namely by graph-based community detection (GCD) algorithms such as Louvain or Leiden, and by k-means.
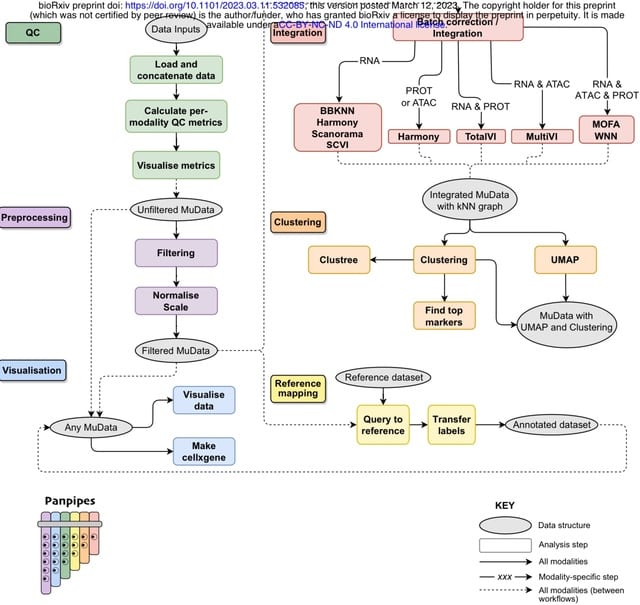
□ Panpipes: a pipeline for multiomic single-cell data analysis.
>>
Panpipes, a set of workflows designed to automate the analysis of multimodal single-cell datasets by incorporating widely used Python-based tools to efficiently perform QC, preprocessing, integration, clustering, and reference mapping at scale in the multiomic setting.
Panpipes generates a cluster matching metric, the Adjusted Rand Index, for global concordance evaluation. Panpipes can aid building unimodal or multimodal references and enables the user to query multiple references simultaneously using scArches.
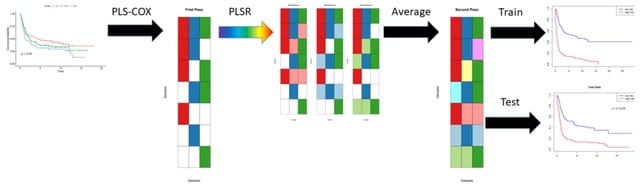
□ plasma: Partial LeAst Squares for Multiomics Analysis
>> https://www.biorxiv.org/content/10.1101/2023.03.10.532096v1
plasma, a novel two-step algorithm to find models that can predict time-to-event outcomes on samples from multiomics data sets even in the presence of incomplete data. These components will be automatically associated with the outcome.
plasma uses partial least squares (PLS) for both steps, using Cox regression to learn the single omics models and linear regression. The plasma components are learned in a way that maximizes the covariance in the predictors and the response.
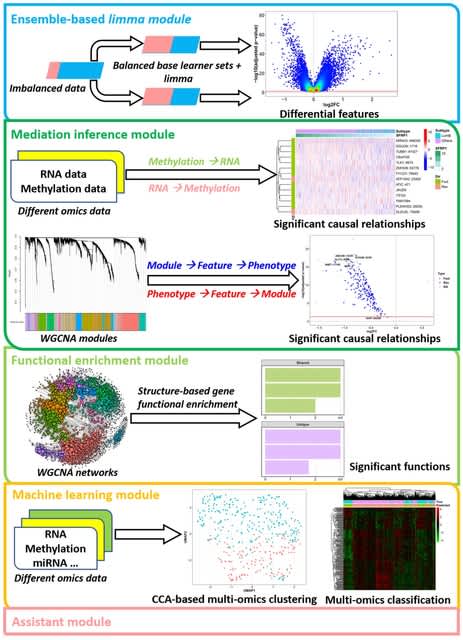
□ eOmics: an R package for improved omics data analysis
>> https://www.biorxiv.org/content/10.1101/2023.03.11.532240v1
eOmics combines an ensemble framework with limma, improving its performance on imbalanced data. It couples a mediation model with WGCNA, so the causal relationship among WGCNA modules, module features, and phenotypes can be found.
eOmics has some novel functional enrichment methods, capturing the influence of topological structure on gene set functions. It contains multi-omics clustering and classification functions to facilitate ML tasks. Some basic functions, such as ANOVA analysis, are also available.

□ Biomappings: Prediction and Curation of Missing Biomedical Identifier Mappings
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad130/7077133
Biomappings, a framework for semi-automatically creating and maintaining mappings in a public, version-controlled repository.
Biomappings combines multiple contributions: (i) a "curation cycle" workflow for creating mappings, (ii) an extensible pipeline for automatically predicting missing mappings between resources, and automatically detecting inconsistencies.
Biomappings currently makes available 9,274 curated mappings and 40,691 predicted ones, providing previously missing mappings between widely used identifier resources covering small molecules, cell lines, diseases, and other concepts.
□ fraguracy: overlapping bases in read-pairs from a fragment indicate accuracy.
>> https://github.com/brentp/fraguracy
Many factors can be predictive of the likelihood of an error. The dimensionality is a consideration because if the data is too sparse, prediction is less reliable. For each combination, while iterating over the bam, it stores the number of errors and the number of total bases in each bin.
fraguracy calculates real error rates using overlapping paired-end reads in a fragment. This avoids some bias. It does limit to the (potentially) small percentage of bases that overlap and it will sample less at the beginning of read 1 and the end of read2.
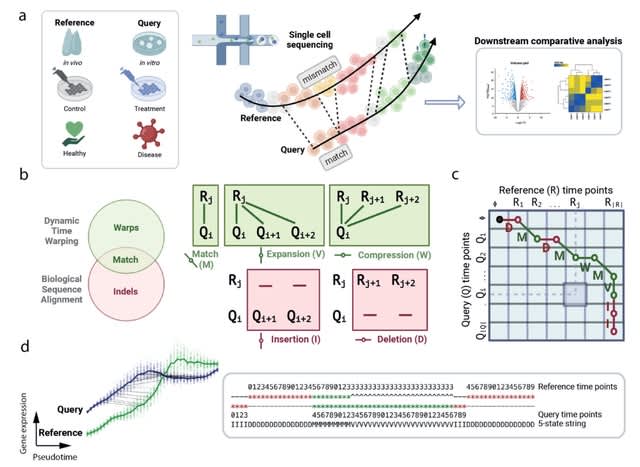
□ Genes2Genes: Gene-level alignment of single cell trajectories informs the progression of in vitro T cell differentiation
>> https://www.biorxiv.org/content/10.1101/2023.03.08.531713v1
Genes2Genes overcomes current limitations and is able to capture sequential matches and mismatches between a reference and a query at single gene resolution, highlighting distinct clusters of genes with varying patterns of gene expression dynamics.
Genes2Genes utilizes a Bayesian information-theoretic Dynamic Programming alignment algorithm that accounts for matches, warps and indels by combining the classical Gotoh’s biological sequence alignment algorithm and Dynamic Time Warping.
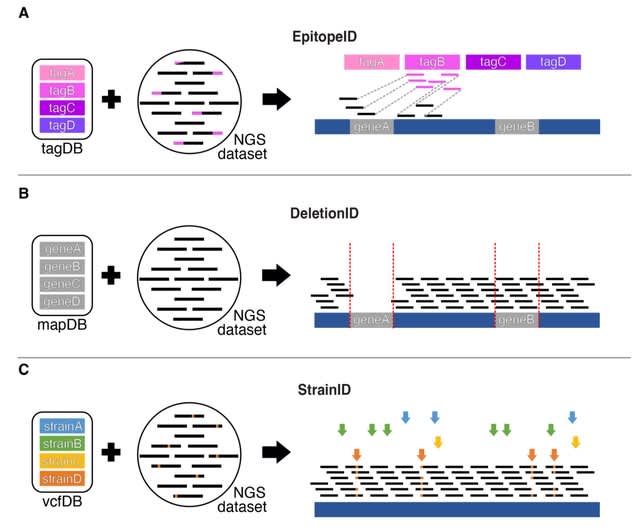
□ GenoPipe: identifying the genotype of origin within (epi)genomic datasets
>> https://www.biorxiv.org/content/10.1101/2023.03.14.532660v1
The three core modules of GenoPipe: EpitopeID, DeletionID, and StrainID were developed to identify major genotypical determinants of cellular identity. GenoPipe can detect genotype perturbations at realistic and practical sequencing depths as defined by ENCODE.
The DeletionID module models the background of a genomic experiment to identify depleted regions of the genome to predict genomic deletions. The StrainID uses existing SNP or variant calls databases of common cell lines to match a cell’s genetic identity inherent to each dataset.
The EpitopeID module identifies the presence and approximate location of specific DNA sequences within the genome. The algorithm functions by first aligning the raw sequencing data (i.e., FASTQ) against a curated DNA sequence database (tagDB) of common protein epitopes.
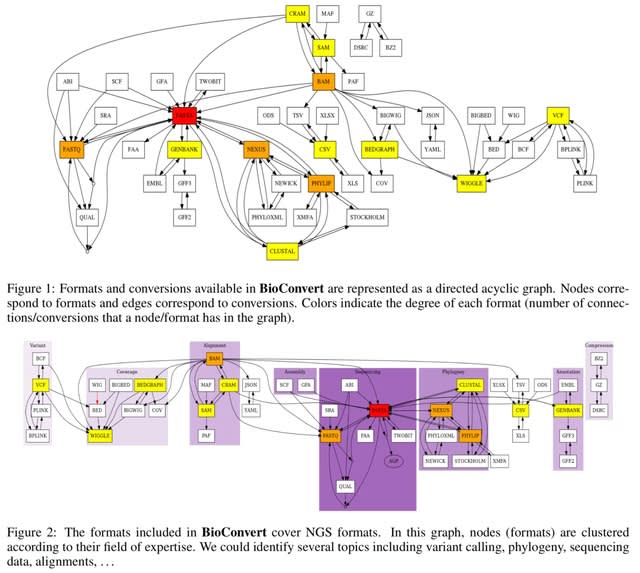
□ BioConvert: a comprehensive format converter for life sciences
>> https://www.biorxiv.org/content/10.1101/2023.03.13.532455v1
BioConvert aggregates existing software within a single framework and complemented them with original code when needed. It provides a common interface to make the user experience more streamlined instead of having to learn tens of them.
BioConvert supports about 50 formats and 100 direct conversions in areas such as alignment, sequencing, phylogeny, and variant calling. BioConvert can also be utilized by developers as a universal benchmarking framework for evaluating and comparing numerous conversion.

□ Fast Approximate IsoRank for Scalable Global Alignment of Biological Networks
>> https://www.biorxiv.org/content/10.1101/2023.03.13.532445v1
A new IsoRank approximation, which exploits the mathematical properties of IsoRank's linear system to solve the problem in quadratic time with respect to the maximum size of the two PPI networks.
A computationally cheaper refinement is proposed to this initial approximation so that the updated result is even closer to the original IsoRank formulation.
In synthetic experiments, they create random graphs using the Erd ̋os R ́enyi and Barab ́asi-Albert models, and ask IsoRank to recover the graph isomorphism between the graphs and a random node permutation.
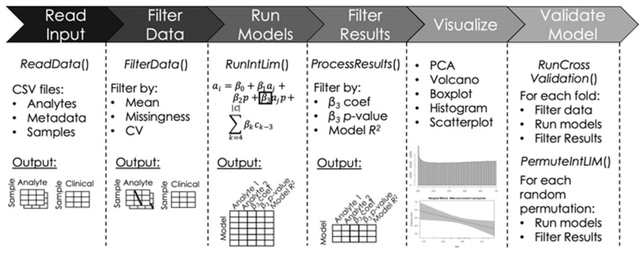
□ IntLIM 2.0: identifying multi-omic relationships dependent on discrete or continuous phenotypic measurements
>> https://academic.oup.com/bioinformaticsadvances/article-abstract/3/1/vbad009/7022005
IntLIM 2.0 uncovers phenotype-dependent linear associations between two types of analytes. IntLIM 2.0 extends IntLIM 1.0 to support generalized analyte measurement data types, continuous phenotypic measurement, covariate correction, model validation and unit testing.
IntLIM 2.0 supports model validation using cross-validation and random permutation models.

□ NanoSquiggleVar: A method for direct analysis of targeted variants based on nanopore sequencing signals
>> https://www.biorxiv.org/content/10.1101/2023.03.15.532860v1
NanoSquiggleVar can directly identify targeted variants from the nanopore sequencing electrical signal without the requirement of base calling, sequence alignment, or variant detection with downstream analysis.
In each sequencing iteration, the signal is sliced into fragments by a moving window of 1-unit step size. Dynamic time warping is used to compare the signal squiggles to the detected variants. NanoSquiggleVar can only determine the existence of a mutation and not its frequency.

□ HiDecon: Accurate estimation of rare cell type fractions from tissue omics data via hierarchical deconvolution
>> https://www.biorxiv.org/content/10.1101/2023.03.15.532820v1
HiDecon, a penalized approach with constraints from both “parent” and “children” cell types to make full use of a hierarchical tree structure. The hierarchical tree is readily available from well-studied cell lineages or can be learned from hierarchical clustering of scRNA-seq.
The basic intuition of HiDecon is that there exists a summation relationship b/n the estimation results of adjacent layers. HiDecon implements the sum constraint penalties from the upper and lower layers to aggregate estimates across layers for more accurate cellular fraction.

□ Implementing Dynamic Time Warping (DTW) with Neural Networks and analyzing single-cell RNA data involves creating a custom model architecture with GPT-4.
□ Yubais RT
昔のAI観ではまず「知性そのもの」みたいなのをコンピュータ内に作って、それと人間が会話するためのインターフェースを別途作るようなイメージだったんだが、インターフェースであるはずの言語に知性っぽいものが内包されていたんじゃないか、と現状を見ていて思う










※コメント投稿者のブログIDはブログ作成者のみに通知されます