









(Paintings by Andrei (@Riabovitchev))
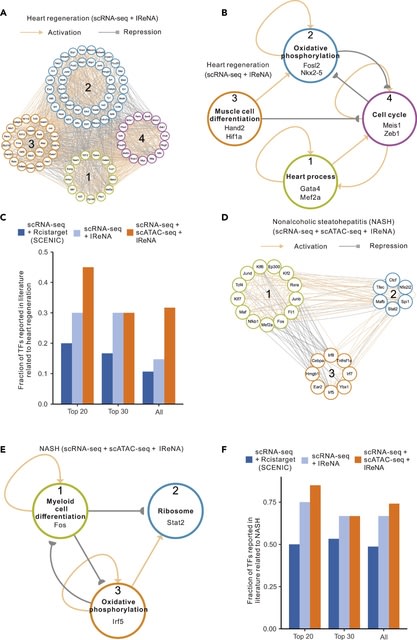
□ IReNA: Integrated regulatory network analysis of single-cell transcriptomes and chromatin accessibility profiles
>> https://www.cell.com/iscience/fulltext/S2589-0042(22)01631-5
Network decoding in IReNA included network modularization, identification of enriched transcription factors, and a unique function for the construction of simplified regulatory networks among modules. Network modularization was based on K-means clustering of gene expression.
IReNA statistically analyzes modular regulatory networks and identifies reliable transcription factors including known regulators. IReNA could directly calculate correlations using original expression data independent of the pseudotime.
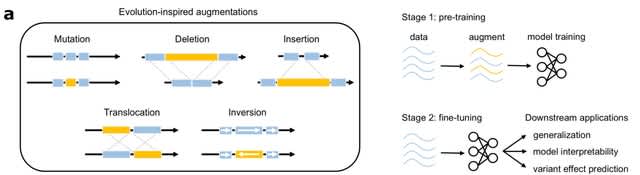
□ EvoAug: Evolution-inspired augmentations improve deep learning for regulatory genomics
>> https://www.biorxiv.org/content/10.1101/2022.11.03.515117v1
EvoAug, an open-source PyTorch package that provides a suite of evolution-inspired data augmentations. EvoAug’s evolution-based augmentations uses the same labels as the original wildtype sequence. This provides a modeling bias to learn invariances of the (un)natural symmetries.
EvoAug randomly applies augmentations, individually or in combinations, online during training to each sequence in a minibatch of data. Each augmentation is applied stochastically and controlled by hyperparameters intrinsic to each augmentation.
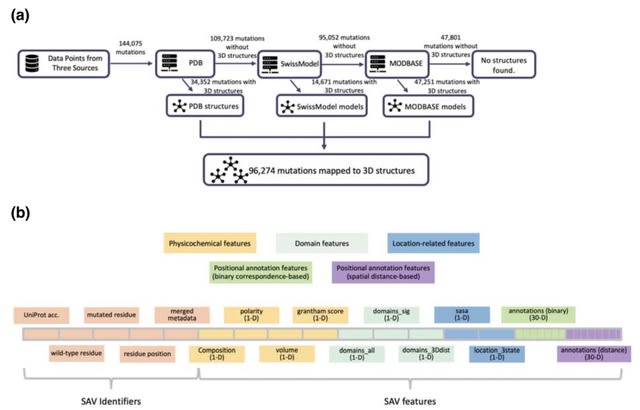
□ ASCARIS: Positional Feature Annotation and Protein Structure-Based Representation of Single Amino Acid Variations
>> https://www.biorxiv.org/content/10.1101/2022.11.03.514934v1
ASCARIS, a method for the featurization (i.e., quantitative representation) of SAVs, which could be used for a variety of purposes, such as predicting their functional effects or building multi-omics-based integrative models.
ASCARIS is incorporated the correspondence between the location of the SAV on the sequence and 30 different types of positional feature annotations. ASCARIS constructed a 74-dimensional feature set to represent each SAV in a dataset composed of ~100,000 data points.
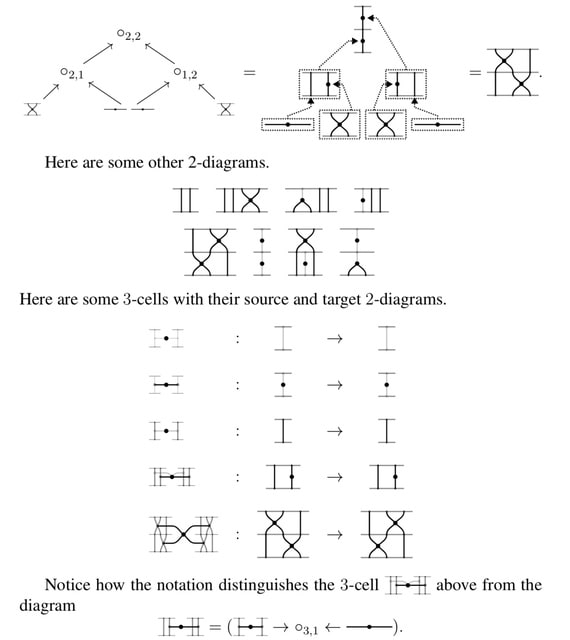
□ Computads and string diagrams for n-sesquicategories
>> https://arxiv.org/pdf/2210.07704.pdf
An n-sesquicategory is an n-globular set with strictly associative and unital composition and whiskering operations, which are however not re-quired to satisfy the Godement interchange laws which hold in n-categories.
The category of computads for this monad is equivalent to the category of presheaves on a small category of computadic cell shapes. Each of these trees has a unique canonical form in its equivalence class.

□ A logical analysis of fixpoint theorems
>> https://arxiv.org/pdf/2211.01782v1.pdf
A fixpoint theorem for Cauchy-complete Q-categories1 that holds for any quantale Q whose underlying complete lattice is continuous and for a specific notion of contraction.
The contractions determine Cauchy distributors under the appropriate algebraic condition on the quantale Q, and finally we formulate the resulting fixpoint theorem for Cauchy-complete Q-categories.
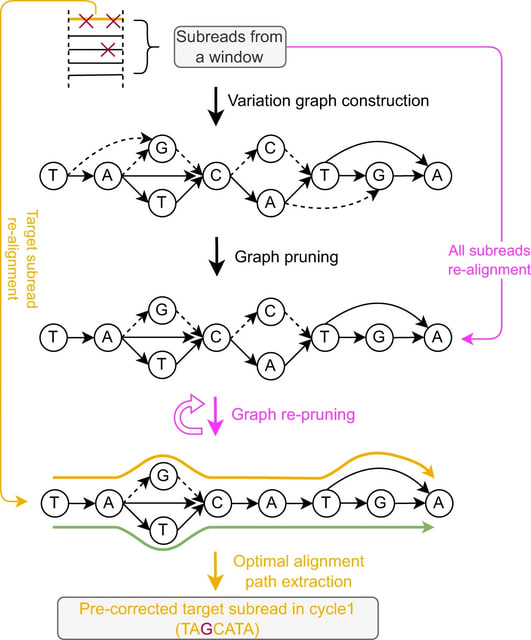
□ VeChat: correcting errors in long reads using variation graphs
>> https://www.nature.com/articles/s41467-022-34381-8
VeChat, a self-correction method to perform haplotype-aware error correction for long reads. VeChat distinguishes errors from haplotype-specific true variants based on variation graphs, which reflect a popular type of data structure for pangenome reference systems.
Unlike single consensus sequences, which current self-correction approaches are generally centering on, variation graphs are able to represent the genetic diversity across multiple, evolutionarily or environmentally coherent genomes.

□ DeepOM: Single-molecule optical genome mapping via deep learning
>> https://www.biorxiv.org/content/10.1101/2022.11.04.512597v1
DeepOM was compared against the state-of-the-art commercial Bionano Solve on human cell-line DNA data acquired with the Bionano Saphyr system. DeepOM enables higher genome coverage from a given sample, enhancing the ability to detect low frequency structural variations.
The DeepOM alignment of a DNA molecule to a reference genome sequence starts from query images of molecules fluorescently labeled at specific motifs. the localization neural network of DeepOM enables the separation of multiple fluorescent emitters that are within a diffraction limited spot.
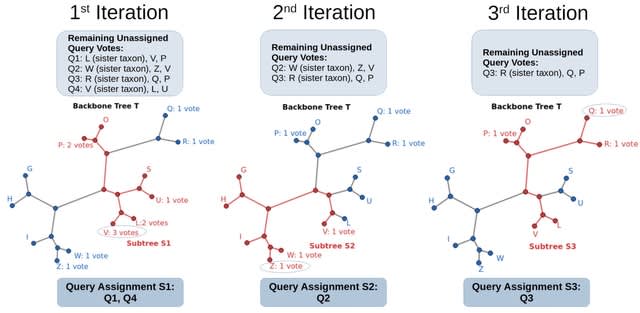
□ BATCH-SCAMPP: Scaling phylogenetic placement methods to place many sequences
>> https://www.biorxiv.org/content/10.1101/2022.10.26.513936v1
BATCH-SCAMPP, a technique that improves scalability in both dimensions: the number of query sequences being placed into the backbone tree and the size of the backbone tree.
BSCAMPP can facilitate the initial tree decomposition of the divide-and-conquer tree estimation pipeline GTM for better placement of shorter, fragmentary sequences into an initial tree containing the longer full-length sequences, potentially leading to final tree estimation.
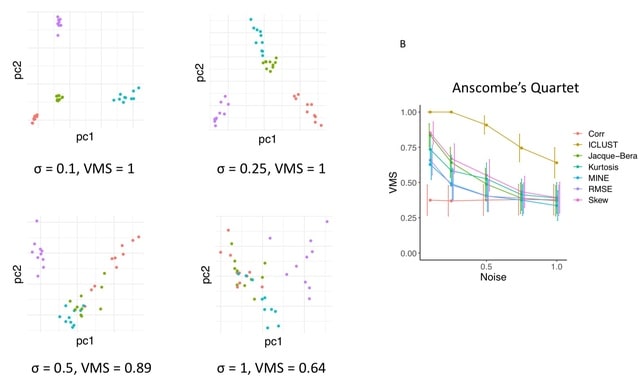
□ ICLUST: Solving Anscombe's Quartet using a Transfer Learning Approach
>> https://www.biorxiv.org/content/10.1101/2022.10.12.511920v1.full.pdf
ICLUST identifies distinct clustering. All scatterplots in the dataset were plotted and clustered using correlation strength alone and 4096-component feature vectors. Average image in each cluster as determined by correlation strength clustering, corresponding to the dendrogram.
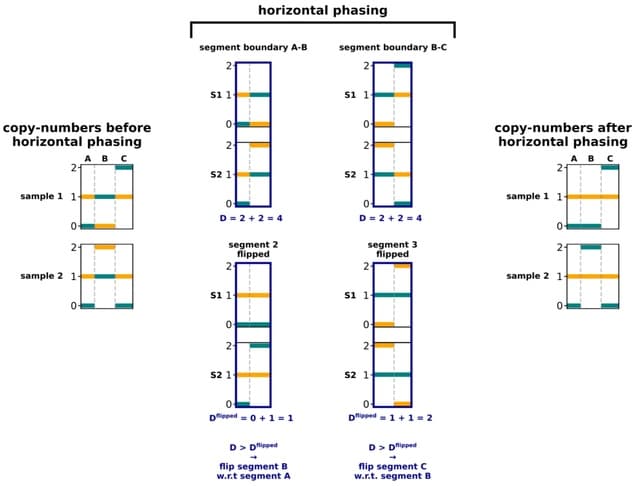
□ Refphase: Multi-sample reference phasing reveals haplotype-specific copy number heterogeneity
>> https://www.biorxiv.org/content/10.1101/2022.10.13.511885v1
Refphase, an algorithm that leverages this multi-sampling approach to infer haplotype-specific copy numbers through multi-sample reference phasing. Unlike statistical phasing, Refphase does not require reference haplotype panels or large collections of genotypes.
Refphase creates a minimum consistent segmentation across the single-sample segmentations input. Allele-specific copy numbers are re-estimated for each sample, and the most parsimonious phasing solution along each chromosome is then chosen in horizontal phasing optimization.
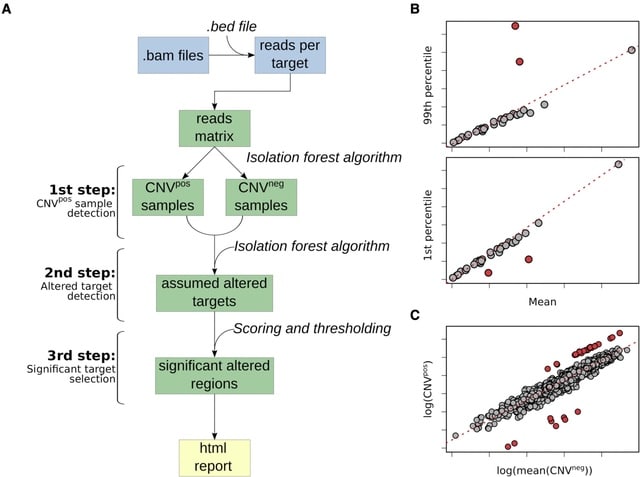
□ ifCNV: A novel isolation-forest-based package to detect copy-number variations from various targeted NGS datasets
>> https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531(22)00252-9
ifCNV is a CNV detection tool based on read-depth distribution obtained from targeted NGS data. ifCNV combines artificial intelligence using two isolation forests and a comprehensive scoring method to faithfully detect CNVs among various samples.
ifCNV integrates a pre-processing step to create a read-depth matrix using as input the aligned bam / bed files. This reads matrix is composed of the samples as columns and the targets as rows. Next, it uses an IF machine learning algorithm to detect the samples w/ a strong bias.
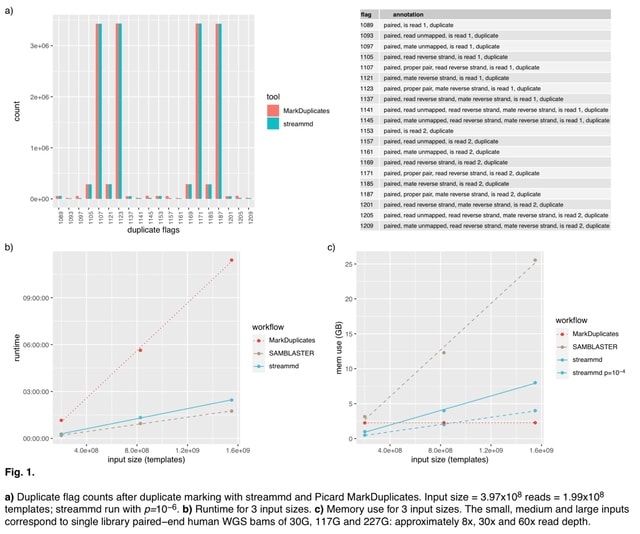
□ streammd: fast low-memory duplicate marking using a Bloom filter
>> https://www.biorxiv.org/content/10.1101/2022.10.12.511997v1
streammd closely reproduces the outputs of Picard MarkDuplicates, a widely-used duplicate marking program, while being substantially faster and suitable for pipelined applications, and that it requires much less memory than SAMBLASTER, another single-pass duplicate marking tool.
With a conventional hash structure the memory requirements of this approach may be considerable for large libraries — a 60x coverage human whole-genome BAM file is around 1B templates and the resulting hash structure tens of GB.
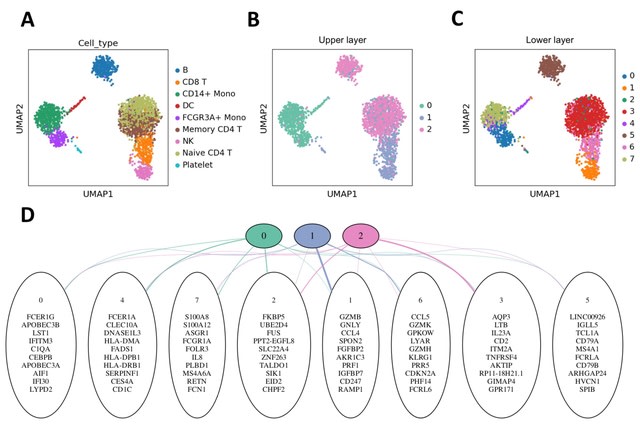
□ scDEF: Deep exponential families for single-cell data analysis
>> https://www.biorxiv.org/content/10.1101/2022.10.15.512383v1
scDEF consists of a deep exponential family model tailored to single-cell data in order to cluster cells using multiple levels of abstraction, which can be mapped to different gene signature levels.
By enforcing non-negativity, biasing towards sparsity and including hierarchical relationships among factors without using batch annotations, scDEF is a general tool for hierarchical gene signature identification in scRNA-seq data for both single- and multiple-batch scenarios.
scDEF models the gene expression heterogeneity of the cells of a tissue as a set of sparse factors containing gene signatures for different cell states. These factors are related to each other through higher-level factors that encode coarser relationships.
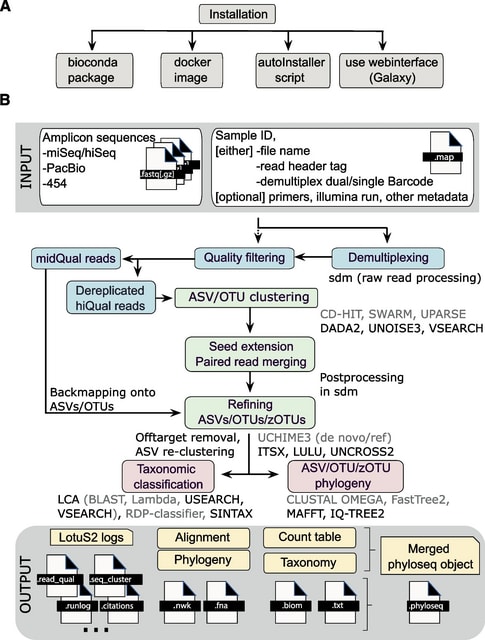
□ LotuS2: an ultrafast and highly accurate tool for amplicon sequencing analysis
>> https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-022-01365-1
LotuS2 is designed to run with a single command, where the only essential flags are the path to input files (fastq(.gz), fna(.gz) format), output directory, and mapping file.
The sequence input is flexible, allowing simultaneous demultiplexing of read files and/or integration of already demultiplexed reads.
The primary output is a set of tab-delimited OTU/ASV count tables, The phylogeny of OTUs/ASVs, their taxonomic assignments, and corresponding abundance tables at different taxonomic levels.
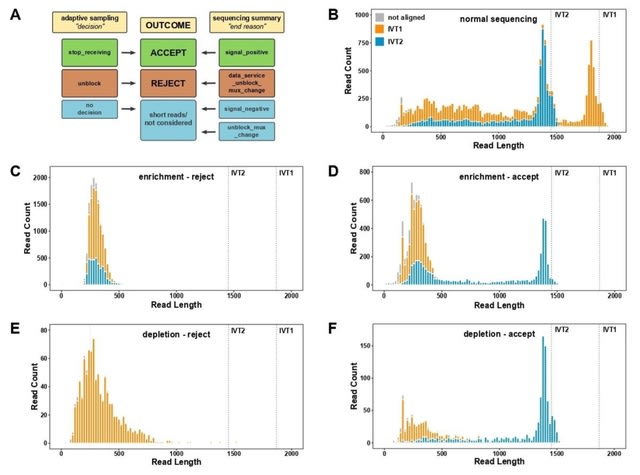
□ Adaptive Sampling as tool for Nanopore direct RNA-sequencing
>> https://www.biorxiv.org/content/10.1101/2022.10.14.512223v1
Taking advantage of a simple model system composed of two defined in vitro transcripts, they determine essential parameters of direct RNA-seq adaptive sampling (DRAS).
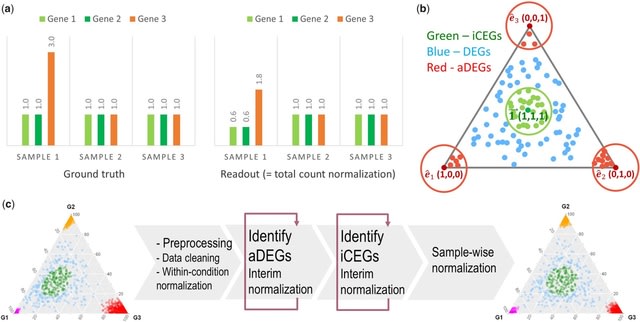
□ Cosbin: cosine score-based iterative normalization of biologically diverse samples
>> https://academic.oup.com/bioinformaticsadvances/article/2/1/vbac076/6764617
A Cosine score-based iterative normalization (Cosbin) method that eliminates aDEGs, identifies ideal CEGs (iCEGs) and calculates sample-wise normalization factors by equilibrating expression levels of iCEGs.
Impactful aDEGs with higher scores are sequentially identified and removed then interim normalization is performed by equilibrating expression levels for the remaining genes, and Cosbin iterates to the next round of aDEG identification and interim normalization.
Sequential elimination of impactful aDEGs should ease the asymmetry in differential expression, reduce normalization bias and improve the efficiency of identifying the next aDEG. Iterations continue until aDEG identification or interim normalization converges at a stable point.
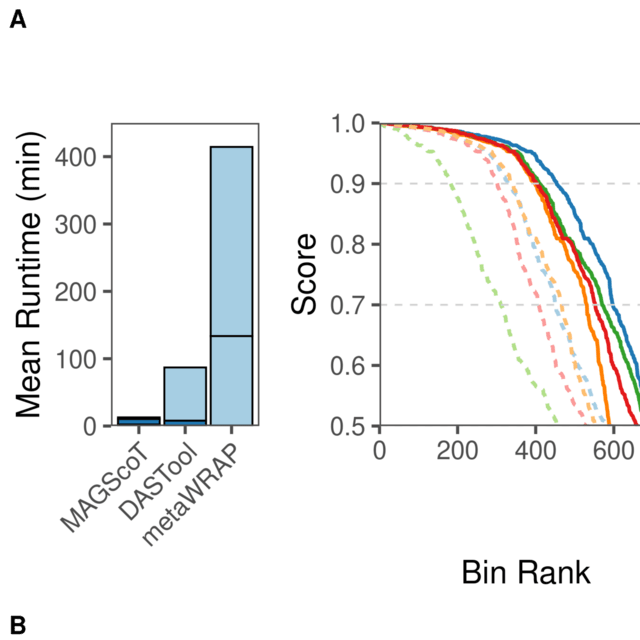
□ MAGScoT - a fast, lightweight, and accurate bin-refinement tool
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac694/6764585
MAGScoT relies on two sets of microbial single-copy marker genes from the Genome Taxonomy Database Toolkit, 120 bacterial and 53 archaeal, stored as HMM-profiles for fast annotation of amino acid sequences predicted from the assembled contigs.
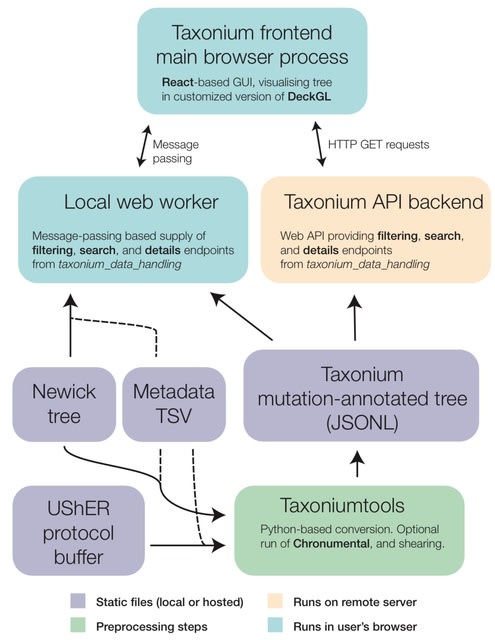
□ Taxonium, a web-based tool for exploring large phylogenetic trees
>> https://www.biorxiv.org/content/10.1101/2022.06.03.494608v4
Taxonium, a new tool that uses WebGL to allow the exploration of trees with tens of millions of nodes in the browser for the first time.
Taxonium links each node to associated metadata and supports mutation-annotated trees, which are able to capture all known genetic variation in a dataset. It can either be run entirely locally in the browser, from a server- based backend, or as a desktop application.
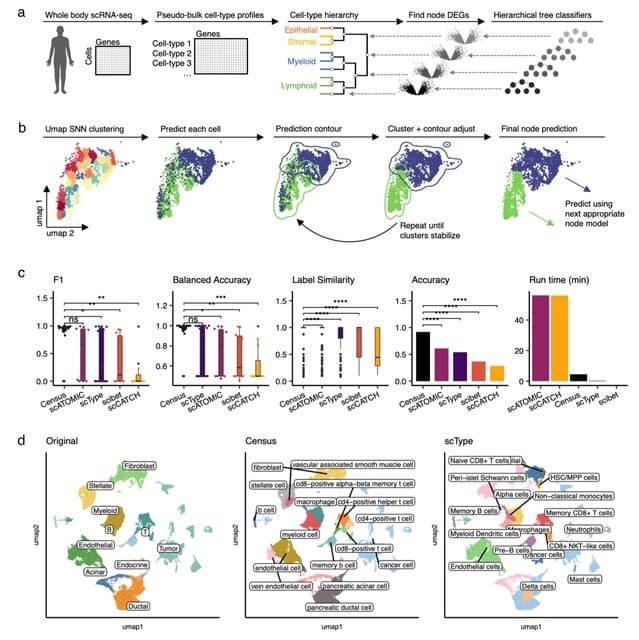
□ Census: accurate, automated, deep, fast, and hierarchical scRNA-seq cell-type annotation
>> https://www.biorxiv.org/content/10.1101/2022.10.19.512926v1
Census implements a collection of hierarchically organized gradient-boosted decision tree models that successively classify individual cells according to a predefined cell hierarchy.
Census begins by identifying a cell-type hierarchy from reference scRNA-seq data by hierarchically clustering pseudo-bulk cell-type gene expression data using Ward’s method, which splits each node into two child nodes.
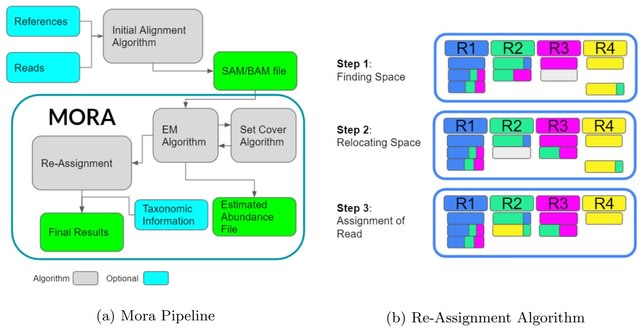
□ Mora: abundance aware metagenomic read re-assignment for disentangling similar strains
>> https://www.biorxiv.org/content/10.1101/2022.10.18.512733v1
Mora is able to accurately re-assign reads by first estimating abundances through an expectation-maximization algo- rithm and then utilizing abundance information to re-assign query reads.
Mora maximizes read re-assignment qualities while simultaneously minimizing the difference from estimated abundance levels, allowing Mora to avoid over assigning reads to the same genomes.

□ DANCE: A Deep Learning Library and Benchmark for Single-Cell Analysis
>> https://www.biorxiv.org/content/10.1101/2022.10.19.512741v1
DANCE platform, the first standard, generic, and extensible benchmark platform for accessing and evaluating computational methods across the spectrum of benchmark datasets for numerous single-cell analysis tasks.
DANCE supports five models for this task. It includes scDeepsort as a GNN-based method. ACTINN and singleCellNet are representative deep learning methods. It also covers support vector machine (SVM) and Celltypist as traditional machine learning baselines.
□ PolyHaplotyper: haplotyping in polyploids based on bi-allelic marker dosage data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04989-0
A new method to reconstruct haplotypes from SNP dosages derived from genotyping arrays, which is applicable to polyploids. This method is implemented in the software package PolyHaplotyper.
PolyHaplotyper is restricted to relatively small haploblocks: in practice the maxima are 8 markers in tetraploids and 6 markers in hexaploids. This theoretically allows to distinguish many different haplotypes, precisely 256 for 8 markers and 64 for 6 markers.
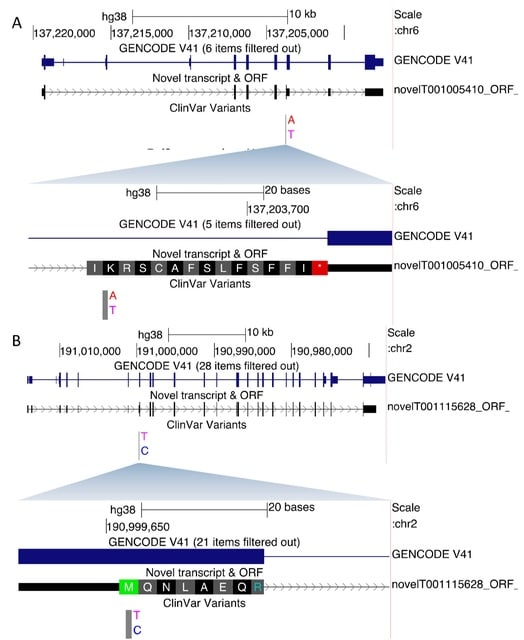
□ SUsPECT: A pipeline for variant effect prediction based on custom long-read transcriptomes for improved clinical variant annotation
>> https://www.biorxiv.org/content/10.1101/2022.10.23.513417v1
SUsPECT (Solving Unsolved Patient Exomes/gEnomes using Custom Transcriptomes), a pipeline based on the Ensembl Variant Effect Predictor (VEP) to predict variant impact on custom transcript sets, such as those generated by long-read RNA-sequencing, for downstream prioritization.
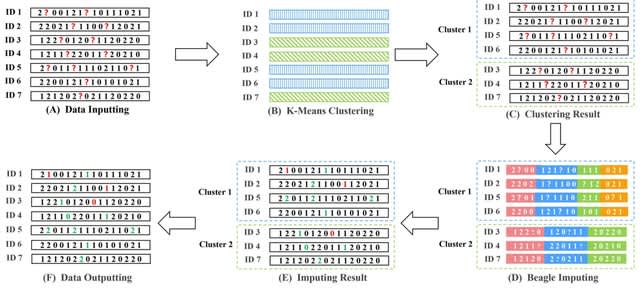
□ KBeagle: An Adaptive Strategy and Tool for Improvement of Imputation Accuracy and Computing Efficiency
>> https://www.biorxiv.org/content/10.1101/2022.10.22.513369v1
Genotype imputation was performed using marker information from the linkage disequilibrium (LD) fragment. The estimated accuracy of fragments between individuals with known and unknown genotypes is the key factor in imputation ability.
KBeagle uses the K-Means algorithm to calculate the genetic distance of samples with missing genotypes, classifying the samples with close genetic distances into one clustered group, and then use the Beagle to estimate the missing genotype of samples in each clustered group.
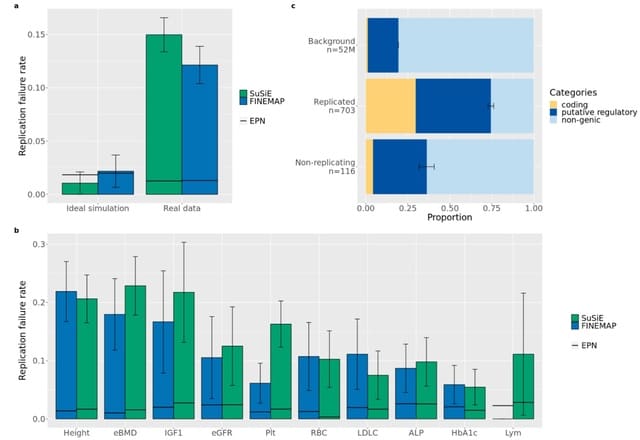
□ RFR: Improving fine-mapping by modeling infinitesimal effects
>> https://www.biorxiv.org/content/10.1101/2022.10.21.513123v1
The Replication Failure Rate (RFR) – a metric that assesses the stability of posterior inclusion probability by evaluating the consistency of PIPs in random subsamples of individuals from a larger well-powered cohort – in this instance for 10 quantitative traits in the UK Biobank.
the RFR to be higher than expected across traits for several Bayesian fine-mapping methods. Moreover, variants that failed to replicate at the higher sample size were less likely to be coding.
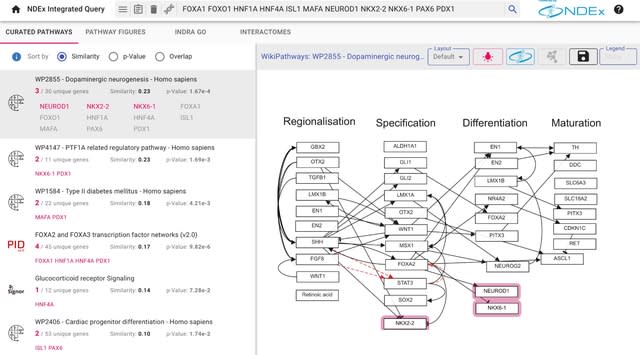
□ NDEx IQuery: a multi-method network gene set analysis leveraging the Network Data Exchange
>> https://www.biorxiv.org/content/10.1101/2022.10.24.513552v1
The NDEx Integrated Query (IQuery) combines novel sources of pathways, integration with Cytoscape, and the ability to store and share analysis results. The IQuery web application performs multiple gene set analyses based on diverse pathways and networks stored in NDEx.
The cosine similarity calculation uses values derived from each gene's term frequency-inverse document frequency (TF-IDF) in the query set and the network. IQuery uses the INDRA system to assemble the output of multiple automated literature mining systems.
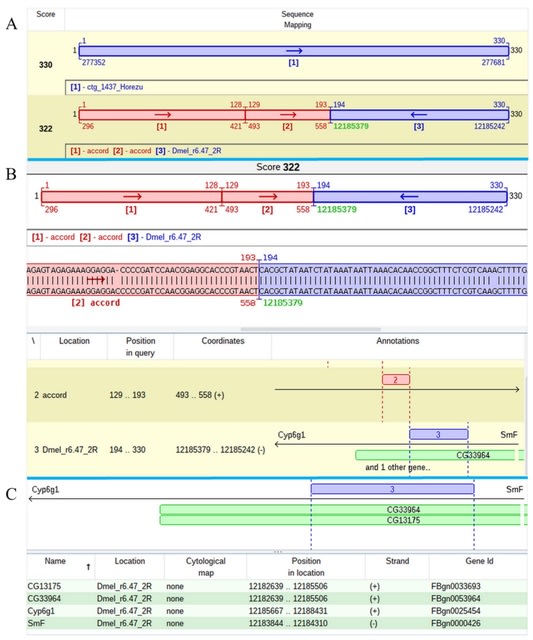
□ Genome ARTIST_v2-An Autonomous Bioinformatics Tool for Annotation of Natural Transposons in Sequenced Genomes
>> https://www.mdpi.com/1422-0067/23/20/12686
The new functions of GA_v2 qualify it as a tool for the mapping and annotation of natural transposons (NTs) in long reads, contigs and assembled genomes.
The new implemented functions allow users to retrieve subsequences from specific references coordinates without a prior alignment with a query sequence;
To extract a list of target site duplications (TSDs) or of flanking sequences consecutive to the alignment of a set of transposon-genome junction query (JQ) sequences versus reference sequences.
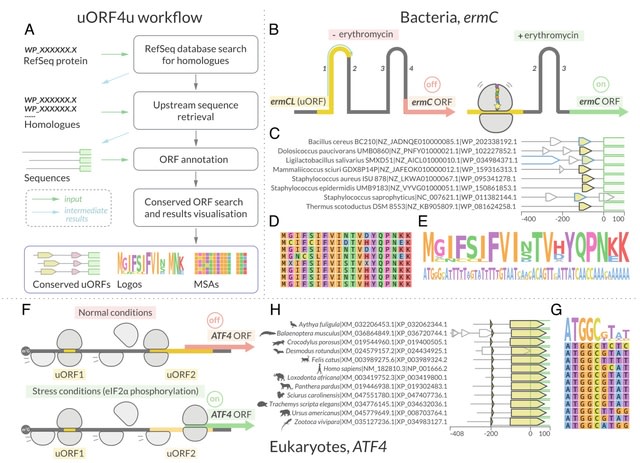
□ uORF4u: a tool for annotation of conserved upstream open reading frames
>> https://www.biorxiv.org/content/10.1101/2022.10.27.514069v1
uORF4u, a tool for conserved uORF annotation in 5ʹ upstream sequences of a user-defined protein of interest or a set of protein homologues. It can also be used to find small ORFs within a set of nucleotide sequences.
If the input is a single RefSeq protein accession number, uORF4u performs a BlastP search against the online version of the RefSeq protein database.
For identified potential frames, the tool searches for conserved ORFs using a greedy algorithm: uORF4u iterates through sequences and tries to maximise the sum of pairwise alignment scores between uORFs.
□ ConsensuSV-from the whole genome sequencing data to the complete variant list
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac709/6782956
The ConsensuSV-core algorithm uses the calls from the individual SV identification algorithms. ConsensuSV starts by preprocessing all the individual VCF files to establish a unified format for further processing.
Every SV is loaded into memory and iterated to find the list of closes ones in terms of their starting position, ending position and type. If the minimum requirement of the number of overlapping candidates is reached, the tool continues processing the list of variants.
□ T1K: efficient and accurate KIR and HLA genotyping with next-generation sequencing data
>> https://www.biorxiv.org/content/10.1101/2022.10.26.513955v1
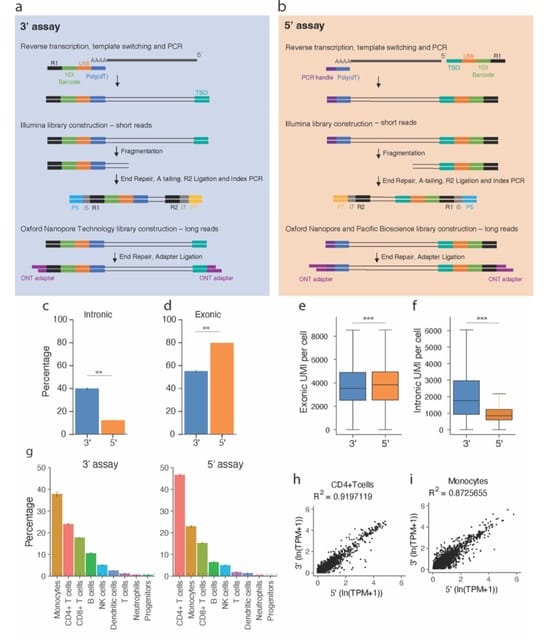
□ Comparing 10x Genomics single-cell 3' and 5' assay in short-and long-read sequencing
>> https://www.biorxiv.org/content/10.1101/2022.10.27.514084v1
Although the barcode detection, cell-type identification, and gene expression profile are similar in both assays, the 5’ assay captured more exonic molecules and fewer intronic molecules compared to the 3’ assay.
13.7% of genes sequenced have longer average read lengths and are more complete (spanning both polyA-site and TSS) in the long reads from the 5’ assay compared to the 3’ assay.
These genes are characterized by long average transcript length, high intron number, and low expression overall. Despite these differences, cell-type-specific isoform profiles observed from the two assays remain highly correlated.
□ Genetic determinism, essentialism and reductionism: semantic clarity for contested science
>> https://www.nature.com/articles/s41576-022-00537-x
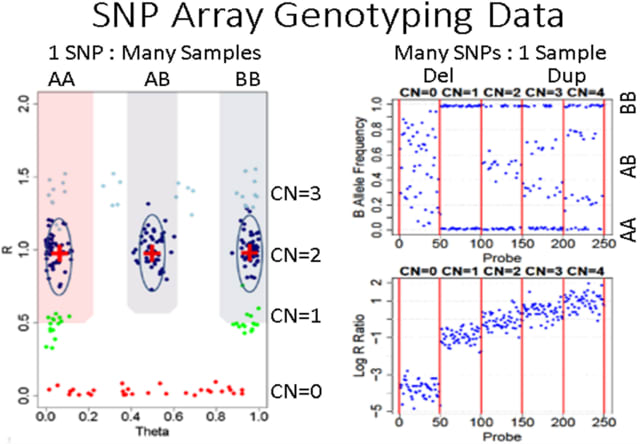
□ ParseCNV2: efficient sequencing tool for copy number variation genome-wide association studies
>> https://www.nature.com/articles/s41431-022-01222-7
ParseCNV2, a next-generation approach to CNV association by natively supporting the popular VCF specification for sequencing-derived variants as well as SNP array calls using a PennCNV format.
ParseCNV2 presents a critical addition to formalizing CNV association for inclusion with SNP associations in GWAS Catalog. Clinical CNV prioritization, interactive quality control (QC), and adjustment for covariates are revolutionary new features of ParseCNV2 vs. ParseCNV.
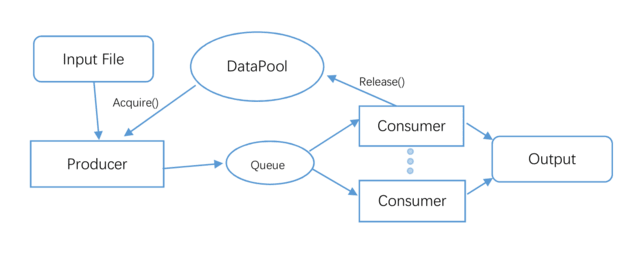
□ RabbitFX: Efficient Framework for FASTA/Q File Parsing on Modern Multi-Core Platforms
>> https://ieeexplore.ieee.org/document/9937043/
RabbitFX can efficiently read FASTA and FASTQ files by combining a lightweight parsing method by means of an optimized formatting implementation.
RabbitFX inegrates three I/O-intensive applications: fastp, Ktrim, and Mash. compared to FQFeeder, in the task of counting ATCG of pair-end data, RabbitFX is 2 times faster in 20 thread.
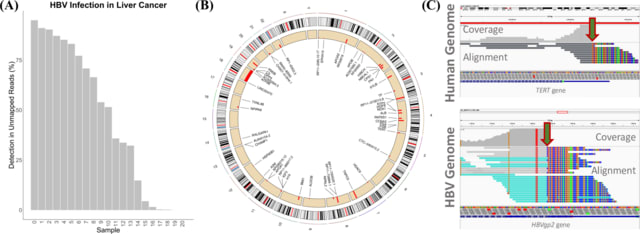
□ Venus: An efficient virus infection detection and fusion site discovery method using single-cell and bulk RNA-seq data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010636
Venus consisted of two main modules: virus detection and integration site discovery. The recommended guideline is to always run the virus detection module but only run the integration module if the virus species is able to integrate its genomic information into the host.
Venus mapped to the integrSeq sequence. Venus classified its chimeric fusion transcripts by biological significance. Venus also ensured that each chimeric read had a clear junction breakpoint, with no gaps or overlaps between the two portions, a quality of true integration sites.
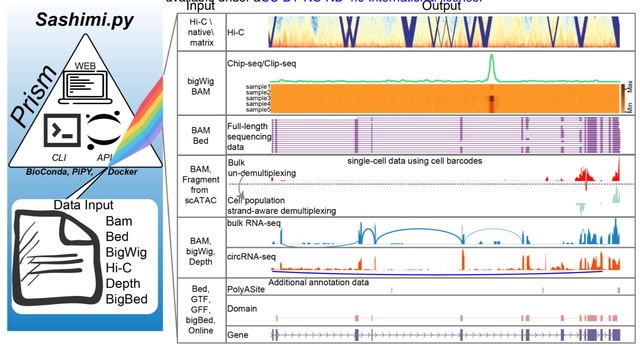
□ Sashimi.py: a flexible toolkit for combinatorial analysis of genomic data
>> https://www.biorxiv.org/content/10.1101/2022.11.02.514803v1
Sashimi.py offers a variety of approaches to use, and users could generate the desired plots by an application programming interface (API) from a script or Jupyter Notebook as well as a command-line interface (CLI).
Sashimi.py is a platform to visually interpret genomic data from a large variety of data sources incl. scRNA-seq, DNA/RNA interactions, long-reads sequencing data, and Hi-C data without any preprocessing, and also offers a broad degree of flexibility for formats of output files.
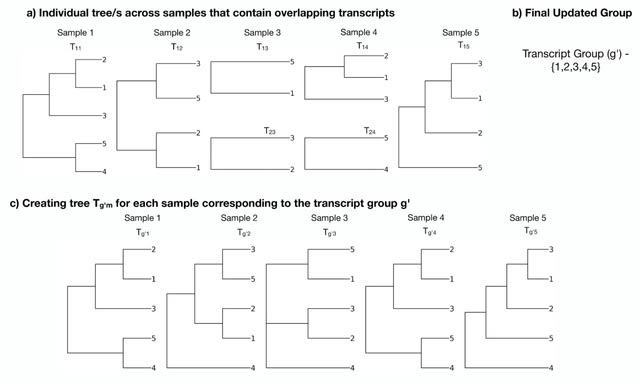
□ TreeTerminus - Creating transcript trees using inferential replicate counts
>> https://www.biorxiv.org/content/10.1101/2022.11.01.514769v1
TreeTerminus, a data-driven approach for grouping transcripts into a tree structure where leaves represent individual transcripts and internal nodes represent an aggregation of a transcript set.
TreeTerminus constructs trees such that, on average, the inferential uncertainty decreases as we ascend the tree topology. TreeTerminus provides a dynamic programming approach that can be used to find a cut through the tree that optimizes one of several different objectives.
□ Proton transfer during DNA strand separation as a source of mutagenic guanine-cytosine tautomers
>> https://www.nature.com/articles/s42004-022-00760-x

□ Entropy: A a visual representation of Entropy increasing on the blockchain. “Absolute Zero”
>> https://opensea.io/collection/entropy-by-nahiko










※コメント投稿者のブログIDはブログ作成者のみに通知されます