








 (photo by Mehran Djo)
(photo by Mehran Djo)
□ Raven: a de novo genome assembler for long reads
>> https://www.biorxiv.org/content/10.1101/2020.08.07.242461v1.full.pdf
Raven is an overlap-layout-consensus based assembler which accelerates overlap step, builds an assembly graph from reads pre-processed, implements a robust simplification method, and polishes the reconstructed contigs Racon, all of which is compiled into a single executable.
Raven searches for suffix-prefix overlaps between the remaining reads enforcing the use of all minimizers. Raven takes 500 CPU hours to assemble a 44x human genome dataset in only 259 fragments.
Raven loads the whole sequencing sample and finds overlaps in fixed-size blocks. Given the quadratic time complexity of the algorithm (O(|V|2)) and 100 iterations until convergence, Raven shrinks the graph by creating unitigs that are 42 vertices away from any junction vertex.

□ AMBER: An automated framework for efficiently designing deep convolutional neural networks in genomics
>>
Automated Modelling for Biological Evidence-based Research (AMBER) is the first automated approach specifically designed for modelling genomic sequences. It leverages the groundbreaking idea of Automated Machine Learning.
AMBER designs optimal models for biological questions through the Neural Architecture Search (NAS). Interpretation of AMBER architecture search revealed its design principles of utilizing the full space of computational operations for accurately modelling genomic sequences.
□ scArches: Query to reference single-cell integration with transfer learning
>> https://www.biorxiv.org/content/10.1101/2020.07.16.205997v1.full.pdf
scArches (single-cell architectural surgery) preserves nuanced biological state information while removing batch effects in the data, despite using four orders of magnitude fewer parameters compared to de novo integration.
scArches is a fast and scalable tool for updating, sharing, and using reference atlases. scArches enables users to share this reference as a trained network with other users, who can in turn update the reference using query-to-reference mapping and partial weight optimization.

□ GLISS: Integrative Spatial Single-cell Analysis with Graph-based Feature Learning
>> https://www.biorxiv.org/content/10.1101/2020.08.12.248971v1.full.pdf
GLISS utilizes a graph-based association measure to select and link genes that are spatially-dependent in both data sources. GLISS can discover new spatial genes and recover cell locations in scRNA-seq data from landmark genes determined from SGE data.
The inference of a one-dimensional temporal relationship shares certain similarities with that of spatial relationships along a one-dimensional latent axis, which is the focus of GLISS.
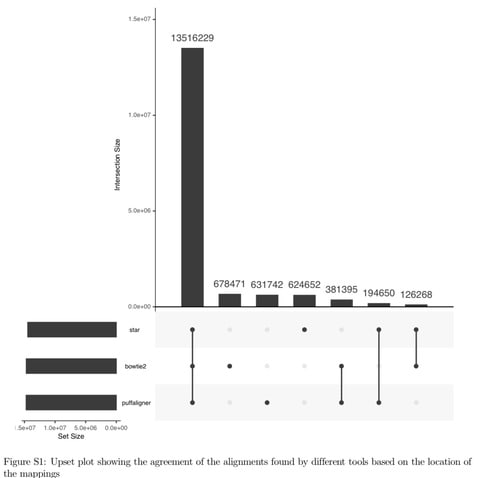
□ Puffaligner: An Efficient and Accurate Aligner Based on the Pufferfish Index
>> https://www.biorxiv.org/content/10.1101/2020.08.11.246892v1.full.pdf
Puffaligner is based on hashing relatively long seeds and then extending them to MEMs, and so it is very fast (typically much faster than approaches based on arbitrary pattern matching in the BWT). It takes a seed - chain - align approach similar to BWA-MEM and minimap2.
Puffaligner tries to occupy a less-well-explored position in the space of read aligners, typically using more memory than BWT-based approaches (unless there are highly repetitive references), but considerably less than very fast but memory-hungry aligners like STAR.

□ ARPEGGIO: Automated Reproducible Polyploid EpiGenetic GuIdance workflOw
>> https://www.biorxiv.org/content/10.1101/2020.07.16.206193v1.full.pdf
the Automated Reproducible Polyploid EpiGenetic GuIdance workflOw (ARPEGGIO) includes all the steps from raw WGBS data to a list of genes showing differential methylation: conversion check, quality check, trimming, alignment, read classification, methylation extraction, statistical analysis and downstream analysis.
ARPEGGIO utilizes an updated read classification algorithm (EAGLE-RC) that supports bisulfite-treated reads and does not require variant information between subgenomes.

□ MESSI: Identifying signaling genes in spatial single cell expression data
>> https://www.biorxiv.org/content/10.1101/2020.07.27.221465v1.full.pdf
Mixture of Experts for Spatial Signaling genes Identification (MESSI) relies on multi-task learning using information from neighboring cells to improve the prediction of response genes within a cell.
the MESSI model uses as input a subset of inter-/intra- signaling genes to predict the expression of a set of response genes. The use of multi-task learning further enables the sharing of information among response genes via joint learning of response genes’ covariance matrices.

□ MAVE-NN: Quantitative Modeling of Genotype-Phenotype Maps as Information Bottlenecks
>> https://www.biorxiv.org/content/10.1101/2020.07.14.201475v1.full.pdf
MAVE-NN currently supports two inference methods: GE regression, which is suitable for datasets with continuous target variables and uniform Gaussian noise, and NA regression, which is suitable for datasets with categorical target variables.
MAVE-NN dramatically reduces the inference time compared to IM regression computed using Metropolis Monte Carlo.
MAVE-NN assumes that, in a MAVE experiment, the underlying G-P map first compresses an input sequence into a single meaningful scalar – the latent phenotype – and that this quantity is read out only indirectly by a noisy and nonlinear measurement process.
□ TALC: Transcript-level Aware Long Read Correction
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btaa634/5872522
TALC (Transcript-level Aware Long Read Correction changes in RNA expression and isoform representation in a weighted De-Bruijn graph to correct long reads.
Transcript-level aware correction by TALC improves the accuracy of the whole spectrum of downstream RNA-seq applications and is thus necessary for transcriptome analyses that use long read technology.
□ Bedtk: Finding Interval Overlap with Implicit Interval Tree
>> https://www.biorxiv.org/content/10.1101/2020.07.07.190744v1.full.pdf
Efficiently finding overlapping intervals is a core functionality behind all interval processing tools. While this strategy improves performance, it is less convenient to use and is limited to a subset of interval operations.
bedtk, a new toolkit for manipulating genomic intervals in the BED format. It supports sorting, merging, intersection, subtraction and the calculation of the breadth of coverage. Bedtk employs implicit interval tree, a new data structure for fast interval overlap queries.
□ qSNE: Quadratic rate t-SNE optimizer with automatic parameter tuning for large data sets
>> https://academic.oup.com/bioinformatics/article/doi/10.1093/bioinformatics/btaa637/5871347
qSNE uses a quasi-Newton optimizer, allowing quadratic convergence rate, and automatic perplexity (level of detail) optimizer. qSNE can fully utilize parallelization at both vector instruction and thread levels.
qSNE requires an order of magnitude fewer iterations for convergence, but on the other hand the cost per iteration is slightly larger by a constant factor if the Hessian matrix rank is O(1), and to be insignificant even when considering an equal number of iterations.
□ Automated assembly of centromeres from ultra-long error-prone reads
>> https://www.nature.com/articles/s41587-020-0582-4
The analyses reveal putative breakpoints in the manual reconstruction of the human X centromere, demonstrate that human X chromosome is partitioned into repeat subfamilies and provide initial insights into centromere evolution.
the centroFlye algorithm for centromere assembly using long error-prone reads, and apply it to assemble human centromeres on chromosomes 6 and X.
□ Efficient dynamic variation graphs
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btaa640/5872523
libbdsg and libhandlegraph, which use a simple, field-proven interface, designed to expose elementary features of these graphs while preventing common graph manipulation mistakes.
Using a diverse collection of pangenome graphs, these tools allow for efficient construction and manipulation of large genome graphs with dense variation.

□ Domino: reconstructing intercellular signaling dynamics with transcription factor activation in model biomaterial environments
>> https://www.biorxiv.org/content/10.1101/2020.07.24.218537v1.full.pdf
Creating an “atlas” with data from a large number of cells may not be adequate to accurately define physiological properties or therapeutic targets.
Domino generated unique signaling networks and activated cell populations in a large single cell data set from different biomaterial microenvironments that had minimal differential gene expression or cell clustering distribution.
□ GPcounts: Non-parametric modelling of temporal and spatial counts data from RNA-seq experiments
>> https://www.biorxiv.org/content/10.1101/2020.07.29.227207v1.full.pdf
although zero-inflation certainly exists in scRNA-seq data, there may be little benefit in modelling it since the additional zero-inflation parameter can be difficult to identify.
a Gaussian process regression method, GPcounts, implementing negative binomial and zero-inflated negative binomial likelihoods. the naive GP scales cubically with number of time points improved the computational requirements through a sparse inference algorithm from the GPflow library.

□ Hifiasm: Haplotype-resolved de novo assembly with phased assembly graphs
>> https://arxiv.org/pdf/2008.01237.pdf
hifiasm, a new de novo assembler that takes advantage of long high-fidelity sequence reads to faithfully represent the haplotype information in a phased assembly graph.
Unlike other graph-based assemblers that only aim to maintain the contiguity of one haplotype, hifiasm strives to preserve the contiguity of all haplotypes. hifiasm consistently outperforms Falcon and Peregrine which do not take the advantage of exact overlaps.
□ scTyper: a comprehensive pipeline for the cell typing analysis of single-cell RNA-seq data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03700-5
scTyper provides three customized methods for estimating cell-type marker expression, including nearest template prediction (NTP), gene set enrichment analysis (GSEA), and average expression values.
scTyper is comprised of the modularized processes of “QC”, “Cell Ranger”, “Seurat processing”, “cell typing”, and “malignant cell typing”.

□ f5c: GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03697-x
f5c enables DNA methylation detection using nanopore sequencers in real-time (i.e. on-the-fly processing of the output) by using a lightweight embedded computer system equipped with a GPU.
f5c parallelise and optimise an implementation of the dynamic programming algorithm called Adaptive Banded Event Alignment (ABEA) to efficiently run on heterogeneous CPU-GPU architectures.
□ PDR: a new genome assembly evaluation metric based on genetics concerns
>> https://academic.oup.com/bioinformatics/article-abstract/doi/10.1093/bioinformatics/btaa704/5881632
PDR (Pairwise Distance Reconstuction) is a genome assembly evaluation metric. It derives from a common concern in genetic studies, and takes completeness, contiguity, and correctness into consideration. PDRi is a implementation of it by integral.
□ MemorySeq: Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors
>> https://www.cell.com/cell/fulltext/S0092-8674(20)30868-0
MemorySeq combines Luria and Delbrück’s fluctuation analysis with population-based RNA sequencing for identifying genes transcriptome-wide whose fluctuations persist for several divisions.
The identification of non-genetic, multigenerational fluctuations can reveal new forms of biological memory in single cells and suggests that non-genetic heritability of cellular state may be a quantitative property.
□ SVCollector: Optimized sample selection for cost-efficient long-read population sequencing
>> https://www.biorxiv.org/content/10.1101/2020.08.06.240390v1.full.pdf
SVCollector identifies the optimal subset of individuals for resequencing. SVCollector analyzes a population-level VCF file from a low resolution genotyping.
SVCollector implements a fast greedy heuristic and an exact algorithm using integer linear programming. SVCollector will likely also over-represent false positives, which will help with the detection and negative validation of these SV calls.
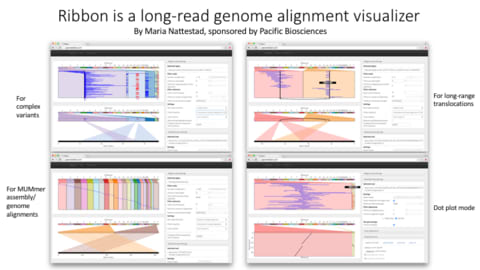
□ Ribbon: Intuitive visualization for complex genomic variation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa680/5885081
Ribbon is an alignment visualization tool that shows how alignments are positioned within both the reference and read contexts, giving an intuitive view that enables a better understanding of structural variants and the read evidence supporting them.
Ribbon was born out of a need to curate complex structural variant calls and determine whether each was well supported by long-read evidence, and it uses the same intuitive visualization method to shed light on contig alignments from genome-to-genome comparisons.

□ LongAGE: defining breakpoints of genomic structural variants through optimal and memory efficient alignments of long reads
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa703/5890675
LongAGE a memory- efficient implementation based on the classical Hirschberg algorithm. an application of LongAGE for resolving breakpoints of SVs embedded into segmental duplications on Pacific Biosciences (PacBio) reads that can be longer than 10Kbp.
LongAGE leverages linear space alignment algorithms based on the idea first presented to solve the longest common subsequence problem and several other such algorithms for sequence alignments.
□ Besca: a single-cell transcriptomics analysis toolkit to accelerate translational research
>> https://www.biorxiv.org/content/10.1101/2020.08.11.245795v1.full.pdf
Besca adds value to bulk RNA-seq studies, especially in larger clinical settings that do not yet have the capacity to perform scRNA- seq and where signals are often confounded by heterogeneity related to distinct cell type composition.
Besca also provides the Besca proportions estimate (Bescape) module, which integrates two cell deconvolution methods: SCDC and MuSiC. And supports analysis of datasets generated by the recently developed CITE-seq, hence accounting for multimodal analysis.
□ ECCO: Efficient and effective control of confounding in eQTL mapping studies through joint differential expression and mendelian randomization analyses
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa715/5892253
ECCO determines the optimal number of PEER factors used for eQTL mapping. Instead of performing repetitive eQTL mapping, ECCO jointly applies differential expression analysis and Mendelian randomization (MR) analysis, leading to substantial computational savings.
ECCO variants are centered around the truth across almost all scenarios, either in the absence of horizontal pleiotropic effects.

□ RabbitQC: High-speed scalable quality control for sequencing data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa719/5892252
RabbitQC mainly focuses on processing uncompressed FASTQ files by a novel I/O-efficient framework. In this framework, the producer thread needs read data from the input file(s) and only processes a few characters of each data chunk.
RabbitQC significantly outperforms oth- ers and achieves about 13x speedup on the 20-core platform. RabbitQC stores a duplication array and a corresponding counting array to provide fast access.

□ GRiNCH: Graph-regularized matrix factorization for reliable detection of topological units from high-throughput chromosome conformation capture datasets
>> https://www.biorxiv.org/content/10.1101/2020.08.17.254615v1.full.pdf
GRiNCH TADs are enriched in known architectural proteins and chromatin modification signals and are stable to the resolution, and sparsity of the input data.
GRiNCH is based on non-negative matrix factorization, a powerful dimensionality reduction method used to recover interpretable low-dimensional structure from high-dimensional datasets. GRiNCH can smooth a sparse input matrix.

□ Information transmission from NFkB signaling dynamics to gene expression
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008011
Analysis of information transmission between ligand and NFkB and ligand and gene expression allows us to determine information loss in transmission between receptors to dynamic signaling patterns and between signaling dynamics to gene expression.
noise-free gene expression has very little information loss suggesting that gene expression can preserve specificity in NFkB patterns. the addition of noise to the gene expression model results in information loss.

□ FuSe: A tool to move RNA-Seq analyses from chromosomal/gene loci to functional grouping of mRNA transcripts
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa735/5894546
To estimate the likelihood of proteins with similar functions, FuSe computes two con- fidence scores: knowledge (KS) and discovery (DS) for protein pairs.
Overlapping protein pairs exhibiting high confidence are grouped to form ‘similar function protein groups’ and expression is calculated for each functional group.
□ nanotatoR: A tool for enhanced annotation of genomic structural variants
>> https://www.biorxiv.org/content/10.1101/2020.08.18.254680v1.full.pdf
OGM-based SV annotation software has seen little development, and currently available SV annotation tools do not provide sufficient information for determination of variant pathogenicity.
nanotatoR provides comprehensive annotation as a tool for SV classification. nanotatoR uses both external (DGV; DECIPHER; Bionano Genomics BNDB) and internal databases to estimate SV frequency.
□ CARE: Context-Aware Sequencing Read Error Correction
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa738/5894969
CARE – an alignment-based scalable error correction algorithm for Illumina data using the concept of minhashing. Minhashing allows for efficient similarity search within large sequencing read collections which enables fast computation of high-quality multiple alignments.


□ Single cell tracking based on Voronoi partition via stable matching
>> https://www.biorxiv.org/content/10.1101/2020.08.20.259408v1.full.pdf
Voronoi partition, a geometric naturalistic method to determine neighbors in a set of objects, and use it as a robust and reliable metric to identify a mappable condition, instead of using the overlap of objects in consecutive frames or nearest distance metric.
□ CLoNe: Automated clustering based on local density neighborhoods for application to biomolecular structural ensembles
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa742/5895303
CLoNe is a clustering algorithm with highly general applicability. Based on the Density Peaks algorithm. CLoNe takes advantage of the Bhattacaryaa coefficient to merge clusters if needed and relies on a Bayes classifier to effectively remove outliers.
CLoNe first performs a Nearest Neighbour step to derive the local densities of every data point. Putative cluster centers are then identified as local density maxima.

□ Dense networks that do not synchronize and sparse ones that do
>> https://aip.scitation.org/doi/10.1063/5.0018322
At the sparse end of the connectivity spectrum, a ring of oscillators can be turned into a globally synchronizing network by adding as few as O(n log2 n) edges in the right places.
Merely connecting each oscillator to a logarithmically small number of neighbors suffices to destabilize all the twisted states of a ring, thereby converting it (we conjecture) into a globally synchronizing network.
□ DataRemix: a universal data transformation for optimal inference from gene expression datasets
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa745/5895302
DataRemix, the simple 3-parameter transformation can be tuned to reweigh the contribution of hidden factors. It can be efficiently optimized via Thompson sampling, which makes it feasible for computationally expensive objectives such as eQTL analysis.
□ reconCNV: Interactive visualization of copy number data from high-throughput sequencing
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa746/5895301
In addition to a standard CNV track for visualizing relative fold change and absolute copy number, reconCNV includes an auxiliary variant allele fraction track for visualizing underlying allelic imbalance and loss of heterozygosity.

□ GBAT: a gene-based association test for robust detection of trans-gene regulation
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02120-1
GBAT uses cvBLUP to produce predictions of gene expression from SNPs cis to each gene. cvBLUP builds leave-one-sample-out cross-validated cis-genetic predictions, to avoid overfitting issues of the standard best linear unbiased predictor.
GBAT reduces false positives caused by RNA-seq alignment errors, by thoroughly removing erroneously mapped RNA-seq reads; multi-mapped reads and reads that are mapped to low mappability regions of the genome, and removing any trans gene pairs that are cross-mappable.

□ EmpiReS: Differential Analysis of Gene Expression and Alternative Splicing
>> https://www.biorxiv.org/content/10.1101/2020.08.23.234237v1.full.pdf
Empirical error distributions for these fold changes are estimated from Replicate measurements and used to quantify feature fold changes and their directions. EmpiReS extends this model such that it can be applied to detect “changes of changes” as is necessary for DAS.
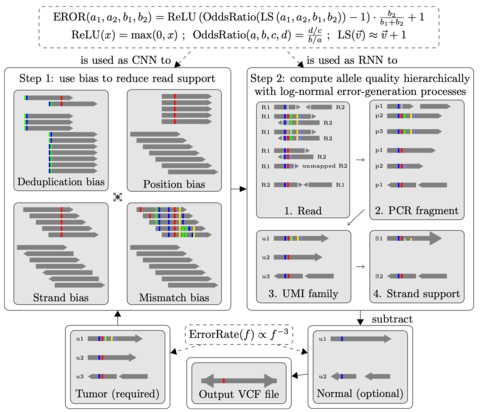
□ UVC: universality-based calling of small variants using pseudo-neural networks
>> https://www.biorxiv.org/content/10.1101/2020.08.23.263749v1.full.pdf
UVC, a Universal and Versatile variant Caller, which utilizes universality and pseudo-neural network (PNN). Pseudo-Neural Network (PNN) resembles a deep neural network in which the weight of each connection between two neurons is predefined to be a mathematical constant such as one. UVC is able to call somatic SNVs and InDels without any prior knowledge.
Power-law model of the relationship b/w allele fraction and false positive probability at infinite depth of coverage. if the coverage depth is high, allele fraction is inversely proportional to the cubic root of variant-calling error probability regardless of variant type.










※コメント投稿者のブログIDはブログ作成者のみに通知されます