










□ SBWT: Succinct k-mer Set Representations Using Subset Rank Queries on the Spectral Burrows-Wheeler Transform
>> https://www.biorxiv.org/content/10.1101/2022.05.19.492613v1.full.pdf
The Spectral Burrows-Wheeler Transform (SBWT) is a distillation of the ideas found in the BOSS and Wheeler graph data structures. The SBWT can also be seen as a specialization of the Wheeler graph framework into k-spectra.
It is possible to use entropy coding methods to compress the space of data structures while retaining query support. MatrixSBWT implemented with bit vectors compressed to the zeroth order entropy leads to a data structure taking 3.25 bits per k-mer on the DNA alphabet.
The space on a general alphabet of size σ is (n+k)(log σ+1/ ln 2)+o((n+k)σ), where n is the number of k-mers in the spectrum. The data structure can answer k-mer membership queries in O(k) time, improving on the BOSS data structure, which occupies the same asymptotic space.
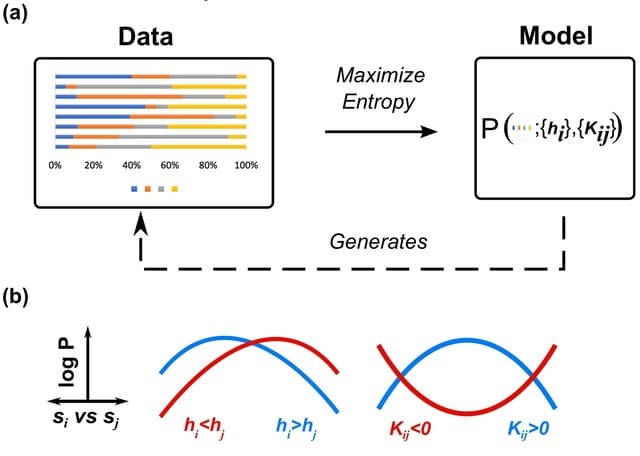
□ The Maximum Entropy Principle For Compositional Data
>> https://www.biorxiv.org/content/10.1101/2022.06.07.495074v1.full.pdf
CME, a data-driven framework for modeling compositions in multi-species networks. CME utilizes maximum entropy, a first-principles modeling approach, to learn influential nodes and their network connections using only the available experimental information.
CME can incorporate more general model constraints as well. The compositional simplex constraint is enforced using the method of Lagrange multipliers. Other geometries, even higher-order moments, can be included simply by including new Lagrange multipliers.
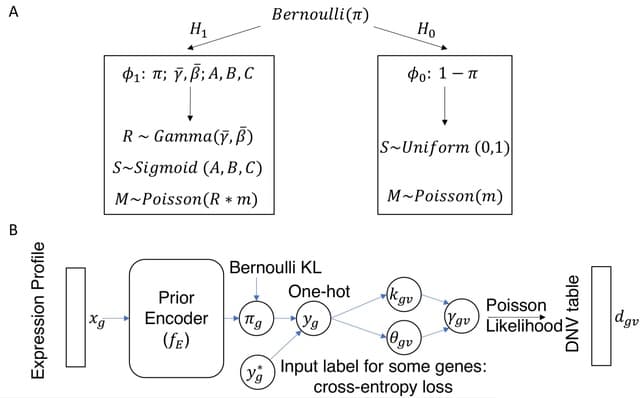
□ xTADA / VBASS: Integration of gene expression data in Bayesian association analysis of rare variants
>> https://www.biorxiv.org/content/10.1101/2022.05.13.491893v1.full.pdf
xTADA takes a single GE profile, such as bulk RNA-seq, as a separate observed variable independent of genetic variants conditioned on risk status. the expression level of a gene is a random variable that has different distributions under the null and the alternative models.
VBASS (Variational inference Bayesian ASSociation), takes a vector of expression profile, and models the priors of risk genes as a function of EP of multiple cell types. VBASS uses deep neural networks to approximate the function and uses semi-supervised variational inference.

□ SPRISS: Approximating Frequent K-mers by Sampling Reads, and Applications
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac180/6588068
SPRISS employs a powerful reads sampling scheme, which allows to extract a representative subset of the dataset that can be used, in combination with any k-mer counting algorithm, to perform downstream analyses in a fraction of the time required by the analysis of the whole data.
SPRISS does not require to receive in input and to scan the entire dataset, but, instead, it needs in input only a small sample of reads drawn from the dataset. the reads-sampling strategy of SPRISS requires the more sophisticated concept of pseudodimension.

□ Exodus: sequencing-based pipeline for quantification of pooled variants
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac319/6584805
Exodus – a reference-based Python algorithm for quantification of genomes, including those that are highly similar, when they are sequenced together in a single mix.
No false negatives were recorded, demonstrating that Exodus’ likelihood of missing an existing genome is very low, even if the genome’s relative abundance is low and similar genomes are sequenced with it in the same mix.
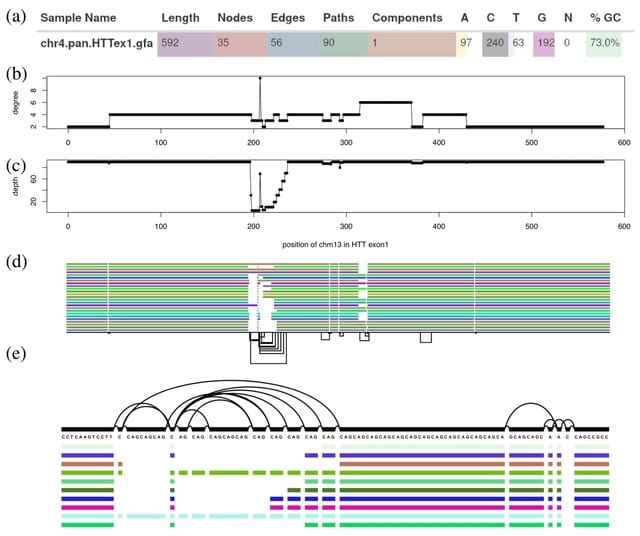
□ ODGI: understanding pangenome graphs
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac308/6585331
ODGI supports pre-built graphs in the Graphical Fragment Assembly format. Its fast parallel execution facilitates routine pangenomic tasks, as well as pipelines that can quickly answer complex biological questions of gigabase-scale pangenome graphs.
ODGI explores context mapping deconvolution of pangenome graph structures via the path jaccard metric. The ODGI data structure allows algorithms that build and modify the graph to operate in parallel, without any global locks.
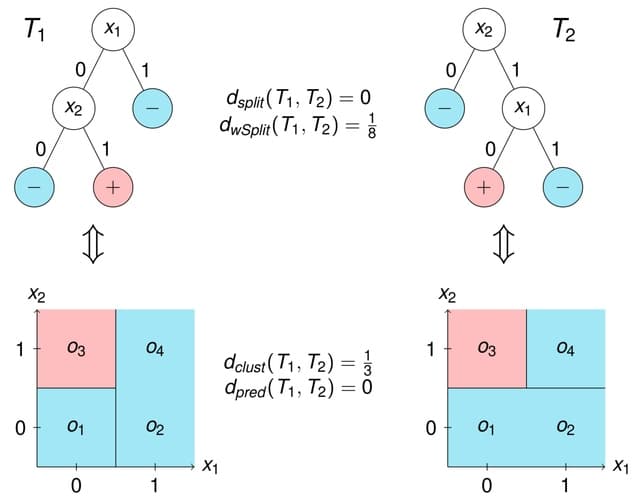
□ WSV: Identification of representative trees in random forests based on a new tree-based distance measure
>> https://www.biorxiv.org/content/10.1101/2022.05.15.492004v1.full.pdf
A new distance measure for decision trees to identify the most representative trees in random forests, based on the selected splitting variables but incorporating the level at which they were selected within the tree.
WSV, the new weighting splitting variable (WSV) metric and describe how to extract the most representative tree from the forest based on any tree distance. WSV approach leads to the best MSE when the minimal node size is small and the trees are therefore more complex.
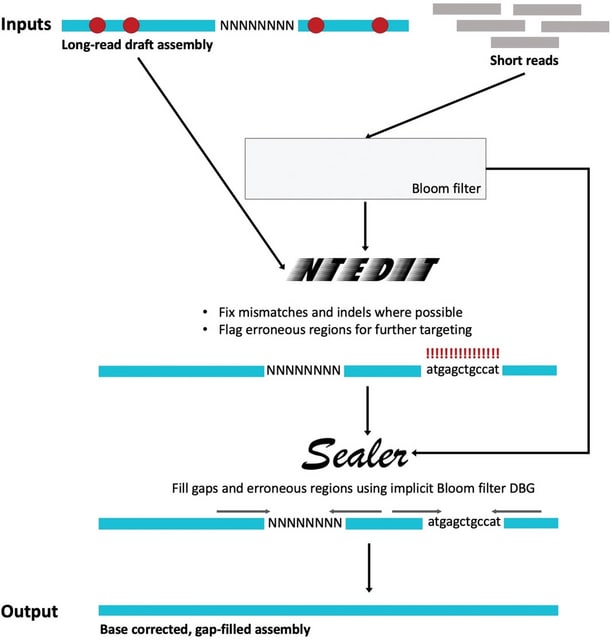
□ ntEdit+Sealer: Efficient Targeted Error Resolution and Automated Finishing of Long-Read Genome Assemblies
>> https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/cpz1.442
ntEdit+Sealer, an alignment-free genome finishing protocol that employs Bloom filters. Both ntEdit / Sealer employ a k-sweep approach, iterating from long to short k-mer lengths. This method is beneficial because different k-mer lengths can provide resolution at different scales.
ntEdit queries assembly k-mers in the Bloom filter, making base corrections where possible and flagging problematic stretches. ABySS-Bloom creates a 2-level cascading Bloom filter, which is used by Sealer as an implicit de Bruijn graph to fill assembly gaps / problematic regions.

□ AtlasXbrowser enables spatial multi-omics data analysis through the precise determination of the region of interest
>> https://www.biorxiv.org/content/10.1101/2022.05.11.491526v1.full.pdf
AtlasXbrowser allows for an assay agnostic image processing GUI that can be used for all DBiT assays. AtlasXbrowser guides the user through the process of locating the region of interest (ROI), defined as the pixels of the micrograph corresponding to the location of the TIXEL mosaic.
AtlasXbrowser encapsulates the numerous advances made in the DBiT protocol since its inception. AtlasXbrowser has standardized the output of DBiT image data, creating a “Spatial Folder”, containing the output of the image processing in the 10x Visium image data format.
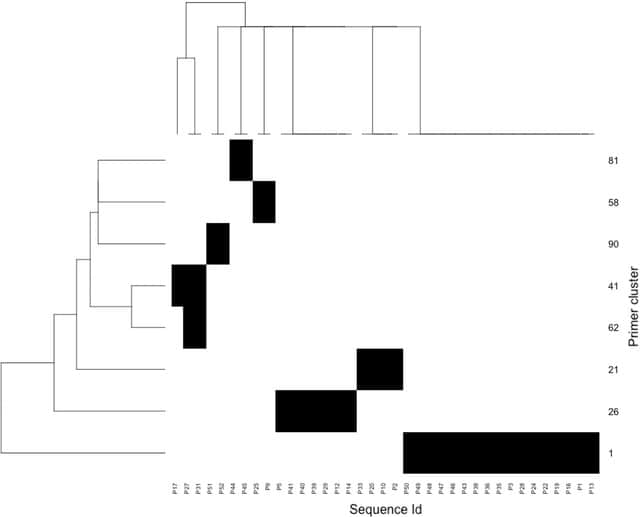
□ Prider: multiplexed primer design using linearly scaling approximation of set coverage
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04710-1
Prider initially prepares a full primer coverage of the input sequences, the complexity of which is subsequently reduced by removing components of high redundancy or narrow coverage.
Prider permits efficient design of primers to large DNA datasets by scaling linearly to increasing sequence data. Prider solves a recalcitrant problem in molecular diagnostics: how to cover a maximal sequence diversity with a minimal number of oligonucleotide primers or probes.
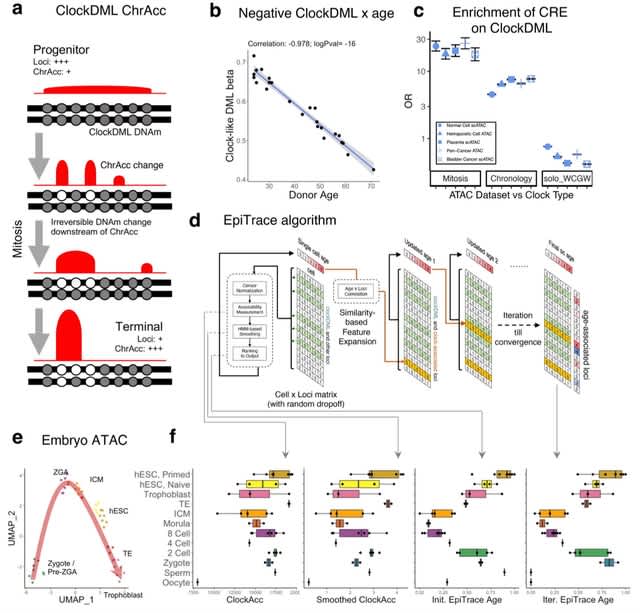
□ EpiTrace: Tracking single cell evolution via clock-like chromatin accessibility
>> https://www.biorxiv.org/content/10.1101/2022.05.12.491736v1.full.pdf
EpiTrace derived cell age shows concordance to known developmental hierarchies, correlates well with DNA methylation-based clocks, and is complementary with mutation-based lineage tracing, RNA velocity, and stemness predictions.
EpiTrace age prediction is reversed for erythroid lineage, probably due to genome-wide chromatin condensation. EpiTrace age shows negative correlation to peaks associated w/ genes acting in current and future stage, and positive correlation to peaks associated with genes acting.
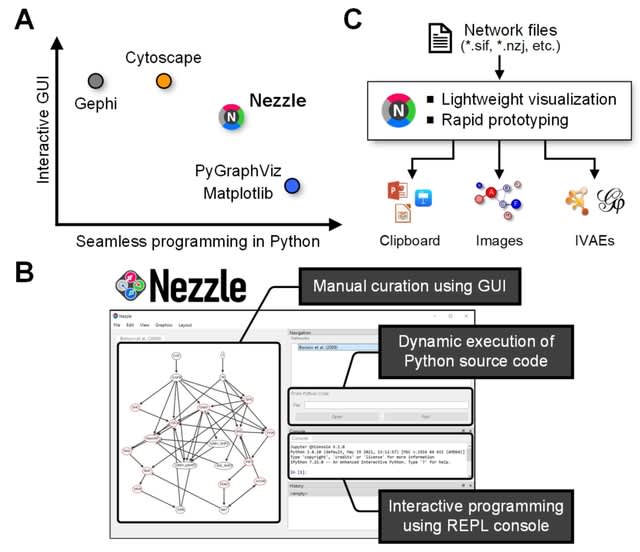
□ Nezzle: an interactive and programmable visualization of biological networks in Python
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac324/6585333
Nezzle provides a set of essential features for rapid prototyping to visualize biological networks. Nezzle provides interfaces for interactive graphics and dynamic code execution.
Nezzle can be a test bed for rapidly evaluating the feasibility of algorithms related to biological networks in Python. Users can develop a prototype of network visualization algorithm that is optimized based on a GPU-accelerated deep learning framework.

□ MultiCens: Multilayer network centrality measures to uncover molecular mediators of tissue-tissue communication
>> https://www.biorxiv.org/content/10.1101/2022.05.15.492007v1.full.pdf
MultiCens (Multilayer/Multi-tissue network Centrality measures) can distinguish within- vs. across-layer connectivity to quantify the “influence” of any gene in a tissue on a query set of genes of interest in another tissue.
MultiCens enjoys theoretical guarantees on convergence and decomposability, and excels on synthetic benchmarks. MultiCens also accounts for the multilayer multi-hop network connectivity structure of the underlying system.
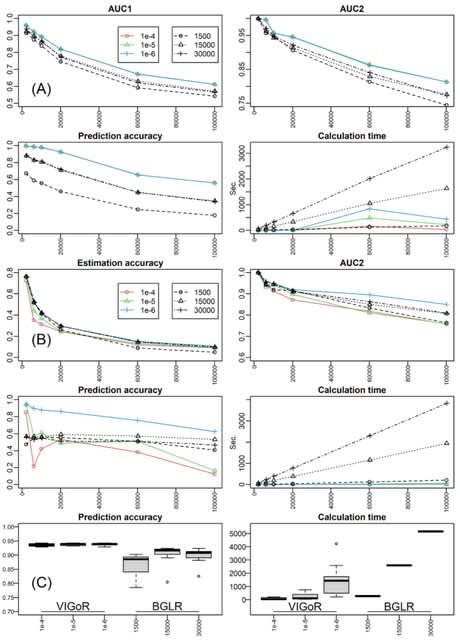
□ VIGoR: joint estimation of multiple linear learners with variational Bayesian inference
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac328/6586288
VIGoR (Variational Bayesian Inference for Genome-Wide Regression) conducts linear regression using variational Bayesian inference, particularly optimized for genome-wide association mapping and whole-genome prediction which use a number of SNPs as the explanatory variables.
Solutions are obtained with variational inference which is more time-efficient than MCMC. VIGoR was initially developed to provide variational Bayesian inference for linear regressions and has been updated to incorporate multiple learners.
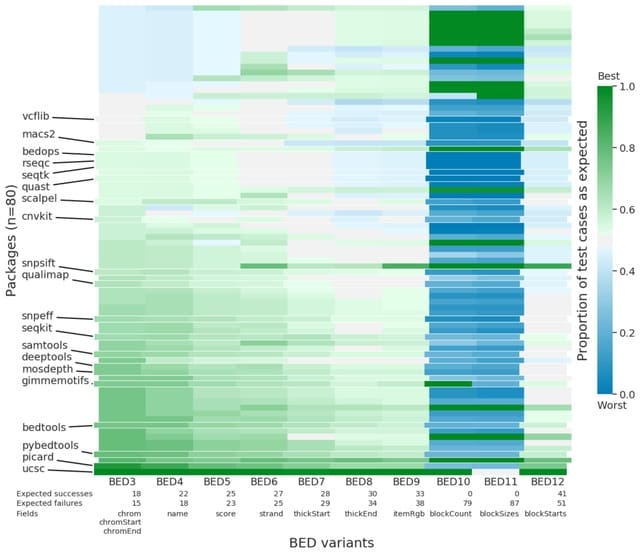
□ Acidbio: Assessing and assuring interoperability of a genomics file format
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac327/6586286
Acidbio, a new verification system which tests for correct behavior in bioinformatics software packages. They crafted tests to unify correct behavior when tools encounter various edge cases—potentially unexpected inputs that exemplify the limits of the format.
BED variants distinguish BED files based on its number of fields. BEDn denotes a file with only the first n fields. BEDn+m denotes a file with the first n fields followed by m fields of custom-defined fields supplied by the user.

□ Building alternative consensus trees and supertrees using k-means and Robinson and Foulds distance
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac326/6586801
A new efficient method for inferring multiple alternative consensus trees and supertrees to best represent the most important evolutionary patterns of a given set of gene phylogenies.
Adapting the popular k-means clustering algorithm, based on some remarkable properties of the Robinson and Foulds distance, can be used to partition a given set of trees into one (for homogeneous data) or multiple (for heterogeneous data) cluster(s) of trees.

□ Comprehensive and standardized benchmarking of deep learning architectures for basecalling nanopore sequencing data
>> https://www.biorxiv.org/content/10.1101/2022.05.17.492272v1.full.pdf
A wide set of evaluation metrics that can be used to analyze the strengths and weaknesses of basecaller models. This toolbox can be used as benchmark for the standardized training and cross-comparison of existing and future basecallers.
Bonito achieved the best overall performance. Using a CRF decoder, over the more traditional CTC decoder, boosts performance significantly and it is likely the reason why Bonito performs so well in the initial benchmark.
Deep RNNs (LSTM) are superior to Transformer layers, and that both simplex and complex convolutional architectures can achieve competitive performance.

□ DCAlign v1.0: Aligning biological sequences using co-evolution models and informative priors
>> https://www.biorxiv.org/content/10.1101/2022.05.18.492471v1.full.pdf
DCAlign returns the ordered sub-sequence of a query unaligned sequence which maximizes an objective function related to the DCA model of the seed. Standard DCA models fail to adequately describe the statistics of insertions and gaps.
DCAlign v1.0 is a new implementation of the Direct Coupling Analysis (DCA) - based alignment technique, DCAlign, which conversely to the first implementation, allows for a fast parametrization of the seed alignment.
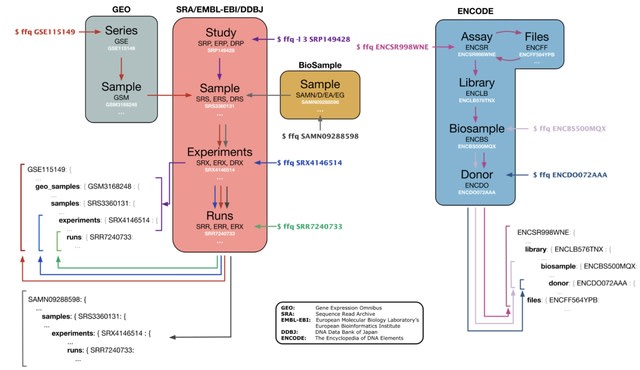
□ ffq: Metadata retrieval from genomics database
>> https://www.biorxiv.org/content/10.1101/2022.05.18.492548v1.full.pdf
ffq facilitates metadata retrieval from a diverse set of databases, including National Center for Biotechnology Information Sequence Read Archive (SRA) and Gene Expression Omnibus (GEO), EMBL-EBI ENA , DDBJ GEA, and ENCODE database.
ffq fetches and returns metadata as a JSON object by traversing the database hierarchy. Subsets of the database hierarchy can be returned by specifying -l [level].
□ SEEM / SEED: Powerful Molecule Generation with Simple ConvNet
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac332/6589886
a ConvNet-based sequential graph generation algorithm. The molecular graph generation problem is reformulated as a sequence of simple classification tasks.
At each step, a convolutional neural network operates on a subgraph that is generated at previous step, and predicts/classifies an atom/bond adding action to populate the input subgraph.
The pretrained model is abbreviated as SEEM (structural encoder for engineering molecules). It is then fine-tuned with reinforcement learning to generate molecules. The fine-tuned model is named SEED (structural encoder for engineering drug-like-molecules).

□ Model verification tools: a computational framework for verification assessment of mechanistic agent-based models
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04684-0
Agent-based models (ABMs) usually make use of pseudo-random number generators initialized with different random seeds for reproducing different stochastic behaviors. It is possible to analyze the model behavior from a deterministic or stochastic point of view.
Model Verification Tools (MVT), a suite of tools based on the same theoretical framework with a user-friendly interface for the evaluation of the deterministic verification of discrete-time models, with a particular focus on agent-based approaches.

□ MATTE: anti-noise module alignment for phenotype-gene-related analysis
>> https://www.biorxiv.org/content/10.1101/2022.05.29.493935v1.full.pdf
A Module Alignment of TranscripTomE (MATTE) aligns modules directly by calculating RDE or RDC transformed data, clustering to assign genes from each phenotype a label, and separating genes into preserved and differentiated modules by cross-tabulation.
MATTE shows a strong anti-noises ability to detect both differential expression and differential co-expression. MATTE takes transcriptome data and phenotype data as inputs hoping to construct a space where each gene from different phenotypes is treated as an individual one.
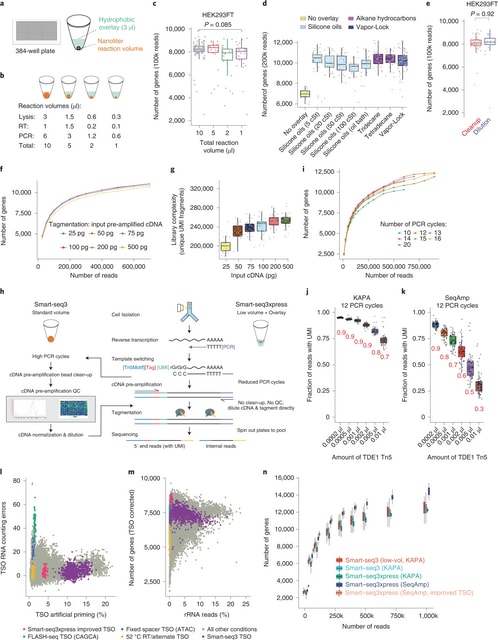
□ Smart-seq3xpress: Scalable single-cell RNA sequencing from full transcripts
>> https://www.nature.com/articles/s41587-022-01311-4
The overlays would both protect the low reaction volumes from evaporation and provide a ‘landing cushion’ for the FACS-sorted cells. Indeed, many overlays with varying chemical properties could be used with low-volume Smart-seq3.
Smart-seq3xpress miniaturizes and streamlines the Smart-seq3 protocol to substantially reduce reagent use and increase cellular throughput. Smart-seq3xpress analysis of peripheral blood mononuclear cells resulted in a granular atlas complete with common and rare cell types.
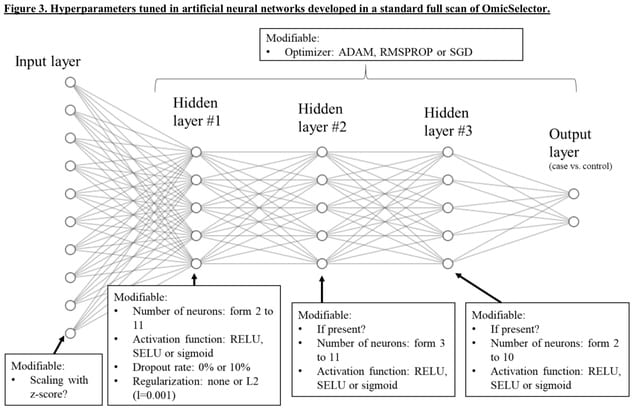
□ OmicSelector: automatic feature selection and deep learning modeling for omic experiments.
>> https://www.biorxiv.org/content/10.1101/2022.06.01.494299v1.full.pdf
OmicSelector provides an overfitting-resilient pipeline that integrates 94 feature selection approaches based on distinct variable selection. OmicSelector identifies the best feature sets using modeling techniques with hyperparameter optimization in hold-out or cross-validation.
OmicSelector provides classification performance metrics for proposed feature sets, allowing researchers to choose the overfitting-resistant biomarker set with the highest diagnostic potential.
OmicSelector performs GPU-accelerated development, validation, and implementation of deep learning feedforward neural networks (up to 3 hidden layers, with or without autoencoders) on selected signatures.

□ DST: Integrative Data Semantics through a Model-enabled Data Stewardship
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac375/6598845
The Data Steward Tool (DST) which can be used to automatically standardize clinical datasets, map them to established ontologies, align them with OMOP standards, and export them to a FHIR-based format.
the DST is capable of automatically mapping external variables onto the CDM through fuzzy string matching. DST provides a graph-based view of the model where the user can interactively explore the entirety of the model.

□ NcPath: A novel tool for visualization and enrichment analysis of human non-coding RNA and KEGG signaling pathways
>> https://www.biorxiv.org/content/10.1101/2022.06.03.494777v1.full.pdf
NcPath integrates a total of 178,308 human experimentally-validated miRNA-target interactions (MTIs), 36,537 experimentally-verified lncRNA target interactions (LTIs), and 4,879 experimentally-validated human ceRNA networks across 222 KEGG pathways.
The NcPath database provides information on MTIs/LTIs/ceRNA networks, PubMed IDs, gene annotations and the experimental verification method used.
the NcPath database will serve as an important and continually updated platform that provides annotation and visualization of the pathways on which noncoding RNAs (miRNA and lncRNA) are involved, and provide support to multimodal noncoding RNAs enrichment analysis.
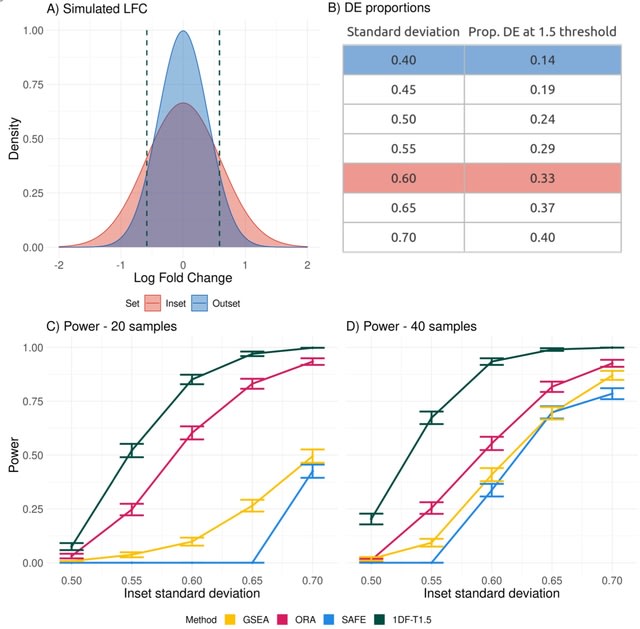
□ Random-effects meta-analysis of effect sizes as a unified framework for gene set analysis
>> https://www.biorxiv.org/content/10.1101/2022.06.06.494956v1.full.pdf
A novel approach to GSA that both provides a unifying framework 39 for the different approaches outlined above and also takes into account the uncertainty in the estimate of the effect size from the first stage of the analysis.
The log fold change (LFC) for genes in a given set is modeled as a mixture of Gaussian distributions, with distinct components corresponding to up-regulated, down-regulated and non-DE genes.
□ ACO:lossless quality score compression based on adaptive coding order
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04712-z
ACO is a special compressor for quality scores, so it considers the distribution characteristics of more quality score data.
The main objective of ACO is to traverse the quality score along the most relative directions, which can be regarded as a reorganization of the stack of independent 1D quality score vectors into highly related 2D matrices.
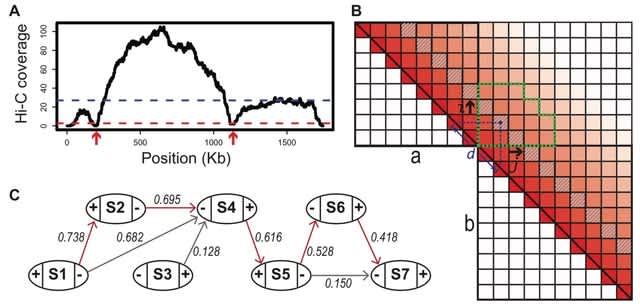
□ YaHS: yet another Hi-C scaffolding tool
>> https://www.biorxiv.org/content/10.1101/2022.06.09.495093v1.full.pdf
YaHS takes the alignment file (either in BED format or BAM format) to first optionally break contigs at positions lacking Hi-C coverage which are potential assembly errors.
YaHS takes account of the restriction enzymes used in the Hi-C library. The cell contact frequencies are normalised by the corresponding number of cutting sites. YaHS builds a scaffolding graph w/ contigs as nodes / contig joins as edges which are weighted by the joining scores.
The graph is simplified by a series of operations incl. filtering low score edges, trimming tips, solving repeats, removing transitive edges, trimming weak edges and removing ambiguous edges. Finally the graph is traversed to assemble scaffolds along contiguous paths.

□ SnapHiC2: A computationally efficient loop caller for single cell Hi-C data
>> https://www.sciencedirect.com/science/article/pii/S2001037022002021
SnapHiC2 adopts a sliding window approach when implementing the random walk with restart (RWR) algorithm, achieving more than 3 times speed up and reducing memory usage by around 70%.
SnapHiC2 can identify 5 Kb resolution chromatin loops with high sensitivity and accuracy. SnapHiC2, with its data-driven strategy to select sliding window size that retains more than 80% of contacts, can identify loops with similar quality as the original SnapHiC algorithm.

□ geometric hashing: Global, highly specific and fast filtering of alignment seeds
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04745-4
Geometric hashing achieves a high specificity by combining non-local information from different seeds using a simple hash function that only requires a constant and small amount of additional time per spaced seed.
Geometric hashing is a fast filter of candidate seeds taken, such as exact k-mer matches induced by spaced seed patterns. the matches from homologous regions are accumulated over possibly long distances. The geometric hashing idea generalizes well for higher dimensional seeds.
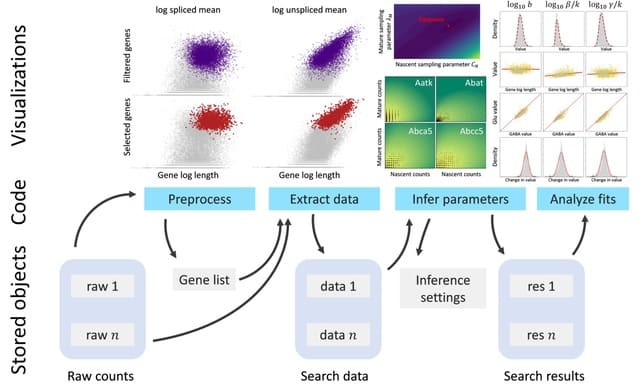
□ Monod: mechanistic analysis of single-cell RNA sequencing count data
>> https://www.biorxiv.org/content/10.1101/2022.06.11.495771v1.full.pdf
By parameterizing multidimensional distributions with biophysical variables, Monod provides a route to identifying and studying differential expression patterns that do not cause changes in average gene expression.
To account for inter-gene coupling through sequencing, the inference procedure iterates over a grid of technical noise parameters and computes a conditional maximum likelihood estimate (MLE) for each gene’s biological noise parameters.
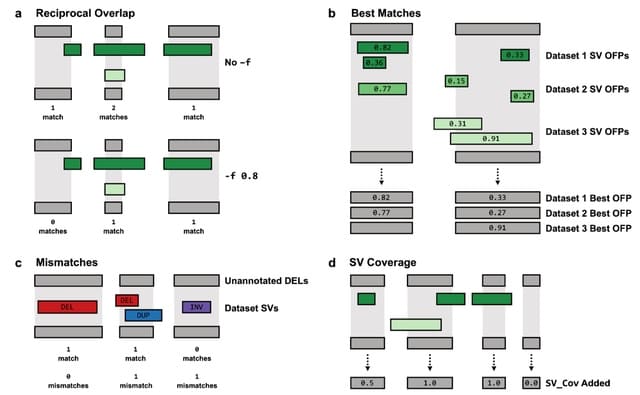
□ SVAFotate: Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets
>> https://www.biorxiv.org/content/10.1101/2022.06.09.495527v1.full.pdf
SVAFotate provides the means to aggregate SV calls from multiple SV population datasets and create summaries of AF-relevant data into simple annotations that are added to SV calls based on default or user-determined SV matching criteria.
SVAFotate has been tested on VCFs created from various SV callers and is compatible w/ any VCF incl. SVTYPE (END / SVLEN) in the INFO field. All SV calls in the VCF are internally converted into a BED for the purposes of identifying overlapping genomic coordinates w/ the SVs.










※コメント投稿者のブログIDはブログ作成者のみに通知されます