










□ One read per cell per gene is optimal for single-cell RNA-Seq:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/09/389296.full.pdf
This approach, although very accurate for deep sequencing, becomes increasingly problematic in the limit of shallow sequencing; overdispersion & inflated dropout levels in lowly expressed genes, typically associated in the literature, are some of the more pronounced consequences. being sensitive to the sequencing depth, significantly overestimates the variability in gene expression due to the inevitable zero-inflation occurring at shallow sequencing, and subsequently limits the performance of common downstream tasks.

□ A Bayesian Approach to Restricted Latent Class Models for Scientifically-Structured Clustering of Multivariate Binary Outcomes:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/25/400192.full.pdf
Conditions ensuring parameter identifiability from the likelihood function are discussed and inform the design of a novel posterior inference algorithm that simultaneously estimates the number of clusters, design matrix Γ, and model parameters. In finite samples and dimensions, we propose prior assumptions so that the posterior distribution of the number of clusters and the patterns of latent states tend to concentrate on smaller values and sparser patterns, respectively. The algorithm adapts the slice sampler for infinite factor model which performs adaptive truncation of the infinite model to finite dimensions and avoids approximation of the Indian Buffet Process (IBP) prior for H∗ under infinite dimension of latent state vectors (ηi).
□ EBADIMEX: An empirical Bayes approach to detect joint differential expression and methylation and to classify samples:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/28/401232.full.pdf
EBADIMEX using empirical Bayes to obtain regularized variance and covariance estimates, generalizing the approach used by limma to multiple dimensions; (1) a moderated Welch t-test for equality of means with unequal variances; (2) a moderated F-test for equality of variances; (3) a multivariate test for equality of means with equal variances.

□ DeepFIGV: Functional Interpretation of Genetic Variants Using Deep Learning Predicts Impact on Epigenome:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/09/389056.full.pdf
DeepFIGV is a deep learning model to accurately predict locus-specific signals from four epigenetic assays using only DNA sequence as input. Given the predicted epigenetic signal from DNA sequence for the reference and alternative alleles at a given locus, DeepFIGV generate a score of the predicted epigenetic consequences for 438 million variants.
□ Implementing a Transcription Factor Interaction Prediction System Using the GenoMetric Query Language:
>> https://link.springer.com/protocol/10.1007/978-1-4939-8561-6_6
GenoMetric Query Language, a novel tool specialized in the integration & management of heterogeneous, large genomic datasets, and a statistical method for robust detection of co-locations across interval-based data, in order to infer physically interacting transcription factors. TICA predictions are supported by existing biological knowledge, making the web server a reliable and efficient tool for interaction screening and data-driven hypothesis generation.
□ OMEGA: An algorithm-centric Monte Carlo method to empirically quantify motion type estimation uncertainty in single-particle tracking:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/08/379255.full.pdf
Even with infinitely accurate and precise positioning, global trajectory measures are nonetheless expected to display statistical variance because of sampling errors (i.e., finite trajectory lengths), which diminishes as the number of points that are available for calculation. Consistently, results presented here and obtained with the OMEGA Diffusivity Tracking Measures plugin, indicate that the accuracy of ODC estimation increases with trajectory length and SNR and starkly depends upon observed ODC and SMSS values of individual trajectories.

□ Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/23/398172.full.pdf
Telescope directly addresses uncertainty in fragment assignment by reassigning ambiguously mapped fragments to the most probable source transcript as determined within a Bayesian statistical model. Telescope performs highly accurate quantification of the retrotranscriptomic landscape in RNA-seq experiments, revealing a differential complexity in the transposable element biology of complex systems not previously observed.

□ An Atlas of Genetic Variation Linking Pathogen-Induced Cellular Traits to Human Disease
>> https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(18)30377-9
Hi-HOST (high-throughput human in vitro susceptibility testing) to identify human genetic differences in pathogen-induced cellular traits, serving as a cell biological link between eQTL studies and GWAS of disease. Hi-HOST uses live pathogens to examine variation in innate immune recognition, but also in pathogen-manipulated cell biological processes that can be quantified as phenotypes for genome-wide association.
□ RNA velocity of single cells
>> https://www.nature.com/articles/s41586-018-0414-6
RNA velocity—the time derivative of the gene expression state—can be directly estimated by distinguishing between unspliced and spliced mRNAs in common single-cell RNA sequencing protocols. RNA velocity is a high-dimensional vector that predicts the future state of individual cells on a timescale of hours. It reveals provides local velocity vectors that can be used to model commitment, fate choice and the precise kinetics of transcription in vivo.
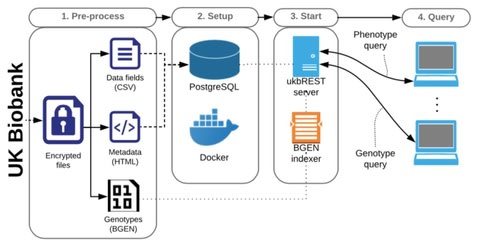
□ ukbREST: efficient and streamlined data access for reproducible research in large biobanks:
>> https://doi.org/10.5281/zenodo.1336815

□ AlleleHMM: a datadriven method to identify allelespecific differences in distributed functional genomic marks:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/10/389262.full.pdf
AlleleHMM uses a hidden Markov model to divide the genome among three hidden states based on allele frequencies in genomic data: a symmetric state 'S' which shows no difference between alleles, and regions with a higher signal on the maternal 'M' or paternal 'P' allele. Using PROseq data, AlleleHMM identified thousands of allele specific blocks of transcription in both coding and noncoding genomic regions.

□ deepMc: deep Matrix Completion for imputation of single cell RNA-seq data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/09/387621.full.pdf
deepMc, a deep Matrix Factorization based imputation technique for scRNA-seq data. its technique does not assume any distribution for gene expression, outperforms other proposed imputation techniques in most experimental conditions, and scales gracefully for a large droplet-sequencing data containing transcriptomes in the order of thousands like PBMCs, having 68K cells.
□ Imperfect Linkage Disequilibrium Generates Phantom Epistasis (& Perils of Big Data):
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/09/388942.full.pdf
the problem of why and under what conditions additive effects may generate “epistatic signals” has not be formalized. In this work, we use a simple three locus model to reveal the conditions that lead to phantom epistasis. if additive QTL variance is imperfectly captured by linear regression on markers and the unexplained variation is not orthogonal to interaction contrasts, then phantom epistasis emerges.

□ CID: High resolution discovery of chromatin interactions:
>> https://www.biorxiv.org/content/biorxiv/early/2018/07/25/376194.full.pdf
CID is more sensitive in discovering chromatin interactions from ChIA-PET data than existing peak-calling-based methods. the improved accuracy and reliability of CID will be important for elucidating the mechanisms of 3D genome folding and long-range gene regulation. With large scale on-going efforts such as the ENCODE project and the 4D Nucleome project, high resolution chromatin interaction mapping from a wider range of tissues and cells will become available in the near future.
□ MetroNome: Organizing genomic data along many dimensions: NYGC's Visualization Tool Aims to Integrate Different Data Types:
>> https://metronome.nygenome.org
MetroNome displays phenotypes in diagrams that attempt to show as much information as possible, applying a technique known as parallel coordinates to all variables that can be expressed numerically.
□ deGSM: memory scalable construction of large scale de Bruijn Graph:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/09/388454.full.pdf
the main idea of deGSM is to efficiently construct the Bur- rows-Wheeler Transformation (BWT) of the unipaths of de Bruijn graph in constant RAM space and transform the BWT into the original unitigs. deGSM is able to handle very large genome sequence(s), e.g., the contigs (305 Gbp) and scaffolds (1.1 Tbp) recorded in Gen-Bank database and Picea abies HTS dataset (9.7 Tbp). DeGSM provides the function to output in GFA (Graphical Fragment Assembly) format, to fulfil the requirements of the emerging graph-based sequence analysis tools.
□ TranslucentID: Detecting Individuals with High Confidence in Saturated DNA SNP Mixtures:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/13/390146.full.pdf
Leveraging differences in DNA contributor concentrations in saturated mixtures, TranslucentID for the identification of a subset of individuals with high confidence who contributed DNA to saturated mixtures by desaturating the mixtures.
□ Magic-BLAST, an accurate DNA and RNA-seq aligner for long and short reads:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/13/390013.full.pdf
Magic-BLAST is the best at intron discovery over a wide range of conditions. It is versatile and robust to high levels of mismatches or extreme base composition and works well with very long reads.
□ cscGANs: Realistic in silico generation and augmentation of single cell RNA-seq data using Generative Adversarial Neural Networks:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/13/390153.full.pdf
cscGANs learn non-linear gene-gene dependencies from complex, multi cell type samples and use this information to generate realistic cells of defined types. The best performing conditional scGAN model (cscGAN) utilized a projection discriminator, along with Conditional Batch Normalization and an LSN function in the generator.
□ Fixation time in evolutionary graphs: a mean field approach:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/14/391508.full.pdf
The method is based on Markov chains and uses a mean field approximation to calculate the corresponding transition matrix. This method can easily be used for a dynamical process with more than two absorption states (for exam- ple a population with more that two types of species) and provides a straightforward tool to calculate all absorption times.
□ scMerge: Integration of multiple single-cell transcriptomics datasets leveraging stable expression and pseudo-replication:
>> http://biorxiv.org/cgi/content/short/393280v1
□ FlowGrid: Ultrafast clustering of single-cell flow cytometry data
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/17/394189.full.pdf
FlowGrid using a new clustering algorithm that combines the advantages of density-based clustering algorithm DBSCAN with the scalability of grid-based clustering. In the multi-centre data sets, FlowGrid shares the similar clustering accuracy (in terms of ARI) with other clustering algorithms but in Seaflow data sets, FlowGrid gives higher accuracy than other clustering algorithms.
□ Linear integral equations, infinite matrices, and soliton hierarchies:
>> https://aip.scitation.org/doi/full/10.1063/1.5046684
A systematic framework is presented for the construction of hierarchies of soliton equations. This is realised by considering scalar linear integral equations and their representations in terms of infinite matrices, which give rise to all (2 + 1)- and (1 + 1)-dimensional soliton hierarchies associated with scalar differential spectral problems. The integrability characteristics for the obtained soliton hierarchies, including Miura-type transforms, τ-functions, Lax pairs, and soliton solutions, are also derived within this framework.
□ On the Number of Driver Nodes for Controlling a Boolean Network to Attractors:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/20/395442.full.pdf
the mathematically prove under a reasonable assumption that the expected number of driver nodes is only O(log2 N + log2 M ) for controlling Boolean networks if the targets are restricted to attractors, where M is the number of attractors.
□ Computational performance and accuracy of Sentieon DNASeq variant calling workflow:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/20/396325.full.pdf
For a WGS sample sequenced to approximately 20X depth, DNASeq can complete the process from FASTQ to VCF in under 2 hours, and from aligned sorted BAM to VCF in less than half an hour. This opens up possibilities for point-of-care patient analysis in the clinic and massive reanalysis of legacy data.
□ DEPECHE: a data-mining algorithm for mega-variate: Determination of essential phenotypic elements of clusters in high-dimensional entities:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/20/396135.full.pdf
DEPECHE, a rapid, parameter free, sparse k-means-based algorithm for clustering of multi- and megavariate single-cell data. In a number of computational benchmarks aimed at evaluating the capacity to form biologically relevant clusters, including flow/mass-cytometry and single cell RNA sequencing data sets with manually curated gold standard solutions.
□ Rust Pseudoaligner:
>> https://github.com/10XGenomics/rust-pseudoaligner
□ LUCA: The last universal common ancestor between ancient Earth chemistry and the onset of genetics:
>> http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1007518
the genetic code and amino acid chirality are universal, all modern life forms ultimately trace back to that phase of evolution. That was the time during which the last universal common ancestor (LUCA) of all cells lived. LUCA is a theoretical constructーit might not have been something we today would call an organism. That approach leads to a different view, that fits well w/ the harsh geochemical setting of early Earth and resembles the biology of prokaryotes that today inhabit the Earth's crust.
□ MSCypher: an integrated database searching and machine learning workflow for multiplexed proteomics.:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/22/397257.full.pdf
MSCypher is a hybrid workflow that currently utilizes the feature detection from the MaxQuant workflow and consists of a combined pre-matching and sensitive search algorithm that interfaces with a supervised machine learning classification using the random forest algorithm. the Andromeda search engine is a natural search algorithm with which to compare and benchmark this software and converting the Andromeda peak lists (APL) and associated information for all features to Mascot generic format (MGF) using the APLtoMGFConverter.
□ SABER enables highly multiplexed and amplified detection of DNA and RNA in cells and tissues:
>> https://www.biorxiv.org/content/biorxiv/early/2018/08/27/401810.full.pdf
Using SABER we were able to detect reporters across a broad range of expression levels, and to assay DNA plasmid copy number in the same cells, providing a tool to quantify enhancer strength and specificity. As an effective and simple method to robustly detect RNA and DNA sequences in cells and tissue, SABER enables the characterization of abundances, identities, and localizations of complex sets of endogenous and introduced nucleic acids.
□ 10x Genomics expands epigenetics offering w/ acquisition of Epinomics & its ATAC-seq platform
Plans to integrate Epinomics IP w/ the Chromium Single Cell ATAC Solution by end of year
>> https://www.10xgenomics.com/










※コメント投稿者のブログIDはブログ作成者のみに通知されます