









人はなぜ物語を必要とするのか。己の置かれた世界に対する仮定、己の取り得る量子的ユニタリーとしての”Trajectries”(軌跡)の複雑性を圧縮し、秩序と混沌の内に自己結晶化する過程に惹き付けられるように共振するからだ。

□ PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1663-x
To trace gene dynamics at single-cell resolution, extended existing random-walk-based distance measures to the realistic case that accounts for disconnected graphs. PAGA covers both aspects of clustering and pseudotemporal ordering by providing a coordinate system (G∗,d) that allows us to explore variation in data while preserving its topology.
PAGA-initialized manifold learning algorithms converge faster, produce embeddings that are more faithful to the global topology of high-dimensional data, and introduce an entropy-based measure for quantifying such faithfulness.
□ STELAR: A statistically consistent coalescent-based species tree estimation method by maximizing triplet consistency
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/31/594911.full.pdf
STELAR (Species Tree Estimation by maximizing tripLet AgReement), an efficient dynamic programming based solution to the CTC problem which is very fast and highly accurate. STELAR runs in O(n^2k|SBP|^2) time.
The algorithmic design in STELAR is structurally similar to ASTRAL.

□ Deep Boltzmann machines : Unsupervised deep learning on biomedical data with BoltzmannMachines.jl
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/20/578252.full.pdf
Deep Boltzmann machines (DBMs) are models for unsupervised learning in the field of artificial intelligence, promising to be useful for dimensionality reduction and pattern detection in clinical and genomic data.
□ SCOUT: A new algorithm for the inference of pseudo-time trajectory using single-cell data
>> https://www.sciencedirect.com/science/article/pii/S1476927119302087
The proposed algorithm is applied to one synthetic and two realistic single-cell datasets (including single-branching and multi-branching trajectories) and the cellular developmental dynamics is recovered successfully.
SCOUT using the projection of Apollonian circle or a weighted distance to determine the pseudo-time trajectories of single cells.

□ Dynverse: A comparison of single-cell trajectory inference methods
>> https://www.nature.com/articles/s41587-019-0071-9
Trajectory inference is unique among most other categories of single-cell analysis methods, such as clustering, normalisation and differential expression, because it models the data in a way that was almost impossible using bulk data.
Dynverse evaluation indicated a large heterogeneity in the performance of the current trajectory inference (TI) methods, with Slingshot, TSCAN, and Monocle DDRTree, towering above all other methods.
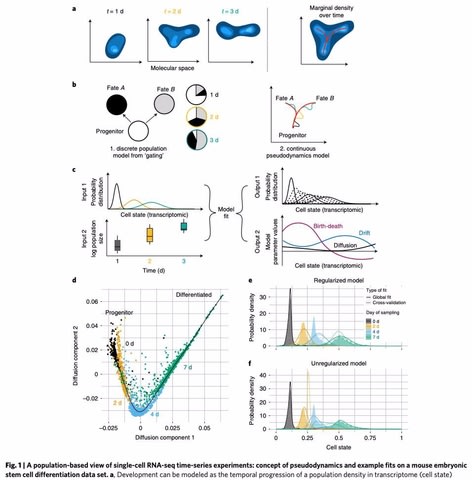
□ Pseudodynamics: Inferring population dynamics from single-cell RNA-sequencing time series data
>> https://www.nature.com/articles/s41587-019-0088-0
pseudodynamics, a mathematical framework that reconciles population dynamics with the concepts underlying developmental trajectories inferred from time-series single-cell data. pseudodynamics adds the following layers of information to a lineage trajectory: model selection between multiple dynamic models such as identification of regions of diffusive and deterministic dynamics.
This model extends previous efforts on modelling gene expression distributions in time by population size dynamics and by the notion of developmental trajectories in transcriptome space.

□ Principal nested shape space analysis of molecular dynamics data
>> https://arxiv.org/pdf/1903.09445.pdf
Principal nested spheres gives a fundamentally different decomposition of data from the usual Euclidean sub-space based PCA. The methodology is applied to cluster analysis of peptides, where different states of the molecules can be identified. Also, the temporal transitions between cluster states are explored.
□ Fundamental Theory of the Evolution Force (FTEF): Gene Engineering utilizing Synthetic Evolution Artificial Intelligence (SYN-AI)
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/21/585042.full.pdf
The effects of the evolution force are observable in nature at all structural levels ranging from small molecular systems to conversely enormous biospheric systems. the evolution force and work associated with formation of biological structures has yet to be described mathematically or theoretically. the driving force of evolution is defined as a compulsion acting at the matter-energy interface that accomplishes genetic diversity while simultaneously conserving structure and function.
According to the “FTEF”, identified genomic building block formations across single and multi-dimension planes of evolution. SYN-AI was able to write functional 14-3-3 ζ docking genes from scratch and present the first theorization and mathematical modelling of the evolution force.

□ FOCUS: Fine-mapping Of CaUsal gene Sets: Probabilistic fine-mapping of transcriptome-wide association studies
>> https://www.nature.com/articles/s41588-019-0367-1
e a probabilistic framework that models correlation among transcriptome-wide association study signals to assign a probability for every gene in the risk region to explain the observed association signal.
FOCUS takes as input summary GWAS data along with eQTL weights and outputs a credible set of genes to explain observed genomic risk.
□ Intelligent Design of 14-3-3 Docking Proteins Utilizing Synthetic Evolution Artificial Intelligence (SYN-AI)
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/23/587204.full.pdf
the DNA tertiary code allows engineering of super secondary structures. SYN-AI constructed a library of 10 million genes that was reduced to three structurally functional 14-3-3 docking genes by applying natural selection protocols.
Synthetic protein identity was verified utilizing Clustal Omega sequence alignments and Phylogeny.fr phylogenetic analysis. Wherein, we were able to confirm three-dimensional structure utilizing I-TASSER and protein ligand interactions utilizing COACH and Cofactor.

□ ASTRAL-MP: scaling ASTRAL to very large datasets using randomization and parallelization
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz211/5418955
ASTRAL uses dynamic programming and is not trivially parallel. ASTRAL-MP, the first version of ASTRAL that can exploit parallelism and also uses randomization techniques to speed up some of its steps.
The ASTRAL-MP code scales very well with increasing CPU cores, and its GPU version, implemented in OpenCL, can have up to 158X speedups compared to ASTRAL-III.
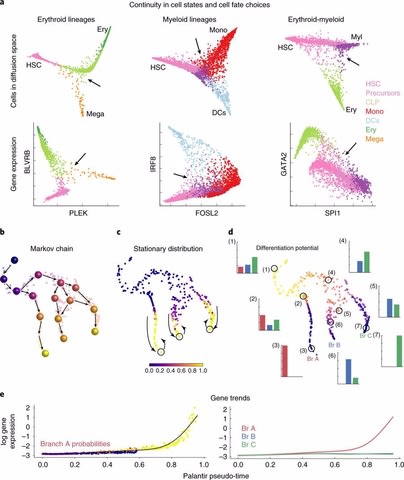
□ Palantir: Characterization of cell fate probabilities in single-cell data
>> https://www.nature.com/articles/s41587-019-0068-4
Palantir is an algorithm to align cells along differentiation trajectories. Palantir models differentiation as a stochastic Markov process where stem cells differentiate to terminally differentiated cells by a series of steps through a low dimensional phenotypic manifold.
Palantir effectively captures the continuity in cell states and the stochasticity in cell fate determination. Palantir has been designed to work with multidimensional single cell data from diverse technologies such as Mass cytometry and single cell RNA-seq.
Palantir generates a high-resolution pseudo-time ordering of cells and, for each cell state, assigns a probability of differentiating into each terminal state.

□ Meta-path Based Prioritization of Functional Drug Actions with Multi-Level Biological Networks
>> https://www.nature.com/articles/s41598-019-41814-w
Meta-paths were utilized to extract the features of each GO term. A meta-path is a sequence of node types and edge types between two nodes at the abstract level.
□ STraTUS: Transmission trees on a known pathogen phylogeny: enumeration and sampling
>> https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msz058/5381076
If only one pathogen lineage can be transmitted to a new host (i.e. the transmission bottleneck is complete), this corresponds to partitioning the nodes of the phylogeny into connected regions, each of which represents evolution in an individual host.
These partitions define the possible transmission trees that are consistent with a given phylogenetic tree. However, the mathematical properties of the transmission trees given a phylogeny remain largely unexplored.

□ Enhancing Boolean networks with continuous logical operators and edge tuning
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/20/584243.full.pdf
The obtained simulations show that continuous results are produced, thus allowing finer analysis. The simulations also show that modulating the signal conveyed by the edges allows to incorporate knowledge about the interactions they model.
The goal is to provide enhancements in the ability of qualitative models to simulate the dynamics of biological networks while limiting the need of quantitative information.
□ Exclusion and genomic relatedness methods for assignment of parentage using genotyping-by-sequencing data
>> https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msz058/5381076
A strategy for using low-depth sequencing data for parentage assignment is developed here. It entails the use of relatedness estimates along with a metric termed excess mismatch rate which, for parent-offspring pairs or trios,
is the difference between the observed mismatch rate and the rate expected under a model of inheritance and allele reads without error. When more than one putative parent has similar statistics, bootstrapping can provide a measure of the relatedness similarity.
□ SAIGE-GENE: Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/20/583278.full.pdf
SAIGE-GENE utilizes state-of-the-art optimization strategies to reduce computational and memory cost, and hence is applicable to exome-wide and genome-wide region- based analysis for hundreds of thousands of samples.
Through the analysis of the HUNT study of 69,716 Norwegian samples and the UK Biobank data of 408,910 White British samples, SAIGE-GENE can efficiently analyze large sample data (N > 400,000) with type I error rates well controlled.

□ Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1667-6
A obust detection of human repeat expansions from careful alignments of long but error-prone (PacBio and nanopore) reads to a reference genome.
This method is robust to systematic sequencing errors, inexact repeats with fuzzy boundaries, and low sequencing coverage. By comparing to healthy controls, prioritize pathogenic expansions within the top 10 out of 700,000 tandem repeats in whole genome sequencing data.

□ A Systems Approach to Refine Disease Taxonomy by Integrating Phenotypic and Molecular Networks
>> https://www.ebiomedicine.com/article/S2352-3964(18)30123-3/fulltext
a new classification of diseases (NCD) by developing an algorithm that predicts the additional categories of a disease by integrating multiple networks consisting of disease phenotypes and their molecular profiles.
With statistical validations from phenotype-genotype associations and interactome networks, NCD improves disease specificity owing to its overlapping categories and polyhierarchical structure.

□ Resolving the full spectrum of human genome variation using Linked-Reads
>> http://m.genome.cshlp.org/content/early/2019/03/20/gr.234443.118.full.pdf
Novel informatic approaches allow for the barcoded short reads to be associated with their original long molecules producing a novel data type known as “Linked-Reads”.
This approach allows for simultaneous detection of small and large variants from a single library. Both Linked-Read whole-genome and whole-exome sequencing identify complex structural variations, including balanced events and single exon deletions and duplications.

□ Chiral DNA sequences as commutable controls for clinical genomics:
>> https://www.nature.com/articles/s41467-019-09272-0
the chiral DNA sequence pairs also perform equivalently during molecular and bioinformatic techniques that underpin genetic analysis, including PCR amplification, hybridization, whole-genome, target-enriched and nanopore sequencing, sequence alignment and variant detection.
□ Prediction of inter-residue contacts with DeepMetaPSICOV in CASP13
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/24/586800.full.pdf
DeepMetaPSICOV (abbreviated DMP), a contact predictor based on a deep, fully convolutional residual network and a large input feature set. DeepMetaPSICOV evolved from MetaPSICOV and DeepCov and combines the input feature sets used by these methods as input to a deep, fully convolutional residual neural network.

□ Can hyperchaotic maps with high complexity produce multistability?
>> https://aip.scitation.org/doi/figure/10.1063/1.5079886
investigate the dynamical behavior in an M-dimensional nonlinear hyperchaotic model (M-NHM), where the occurrence of multistability can be observed.
Four types of coexisting attractors including single limit cycle, cluster of limit cycles, single hyperchaotic attractor, and cluster of hyperchaotic attractors can be found, which are unusual behaviors in discrete chaotic systems. Furthermore, the coexistence of asymmetric and symmetric properties can be distinguished for a given set of parameters.
a simple controller on the M-dimensional nonlinear hyperchaotic model, which can add one more loop in each iteration, to overcome the chaos degradation in the multistability regions.

□ Changes in gene expression shift and switch genetic interactions
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/15/578419.full.pdf
Deep mutagenesis of the lambda repressor reveals that changes in gene expression will alter the strength and direction of genetic interactions between mutations in many genes. A mathematical model that propagates the effects of mutations on protein folding to the cellular phenotype accurately predicts changes in mutational effects and interactions.

□ GenomeWarp: an alignment-based variant coordinate transformation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz218/5420550
The goal of GenomeWarp is to translate the variation within a set of regions deemed "confidently-called" in one genome assembly to another genome assembly.
GenomeWarp transforms regions and short variants in a conservative manner to minimize false positive and negative variants in the target genome, and converts over 99% of regions and short variants from a representative human genome.

□ Developing a network view of type 2 diabetes risk pathways through integration of genetic, genomic and functional data
>> https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-019-0628-8
Genes with cumulative PCS > 0.7 were projected into the InWeb3 dataset using a Steiner tree algorithm to define a PPI network that maximises candidate gene connectivity. This network was further analysed to find processes, pathways and genes implicated in the T2D pathogenesis.
□ Phase space characterization for gene circuit design
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/27/590299.full.pdf
The maintained piecewise linear dynamics across cellular and compositional contexts. Taken together these results show that TU expression dynamics could be predicted by a reference TU up to a context dependent scaling factor.
The combination of TUs and their phase space trajectories reveal the effects of cellular and compositional context on the dynamics of their expression, and suggest approaches for reliable gene circuit design that over-come them.
□ Integer multiplication in time O(n log n)
>> https://hal.archives-ouvertes.fr/hal-02070778
in the multitape Turing model, in which the time complexity of an algorithm refers to the number of steps performed by a deterministic Turing machine with a fixed, finite number of linear tapes. the main results also hold in the Boolean circuit model, with essentially the same proofs.
the theorem implies that quotients and k-th roots of real numbers may be computed to a precision of n significant bits in time O(n log n), and that transcendental functions and constants such as ex and π may be computed to precision n in time O(n log^2 n).
□ edge: The optimal discovery procedure for significance analysis of general gene expression studies
>> https://www.biorxiv.org/content/biorxiv/early/2019/03/27/571992.full.pdf
D dimensional natural cubic spline basis: the knots are placed at evenly spaced quantiles. the basis dimension used for model fitting is chosen by applying a cross validation procedure to select the optimal d across all eigen-genes.

□ KNOT: Knowledge Network Overlap exTraction is a tool for the investigation of fragmented long read assemblies
>> http://pierre.marijon.fr/dow/graph_analysis_of_fragmented_long-read_bacterial_genome_assemblies.pdf
automatically investigate unresolved assemblies and propose directions for refinement, KNOT framework is first tested on synthetic data to illustrate a simple case of fragmentation due to heuristics in the Canu assembler. KNOT recoveres information to provide likely assembly hypotheses using Hamiltonian paths, through a ranked list of contigs orderings.
□ Peregrine: a new OLC assembler:
>> https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/analysis/JasonChin_Peregrine_PacBioCCS_assembly_03212019/README.txt
The Peregrine assembler implements a novel approach for indexing and overlapping reads with a new data structure Sparse and HIierarchical MiniMizER (SHIMMER). The Peregrine overlapper produces overlap file that is compatible to the overlap-to-layout and layout-to-contig modules in the FALCON assembler.
In the current implementation, the read overlapping is finished in less than 7 cpu hours for the 28x coverage dataset of a human genome. Peregrine’s overlap computation time is significantly reduced by using the SHIMMER indexing structure in comparison to other earlier assembly approaches for accurate long reads (length ~ 15kb and error rate < 1%)
□ Linguistics-driven machine learning to decipher the molecular language of immunity (ImmunoLingo)
>> https://www.uio.no/english/research/strategic-research-areas/life-science/research/convergence-environments/immunolingo/index.html
This goal will be achieved by transdisciplinarily combining expertise of life sciences, machine learning, statistics and linguistics researchers. The convergence environment will set out to decipher the molecular language of adaptive immunity, which called ImmunoLingo.

□ BIODICA: Assessing reproducibility of matrix factorization methods in independent transcriptomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz225/5426054
Matrix factorization (MF) methods are widely used in order to reduce dimensionality of transcriptomic datasets to the action of few hidden factors (metagenes). BIODICA for performing the Stabilized ICA-based RBH meta-analysis.
□ Structural variant analysis for linked-read sequencing data with gemtools
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz239/5426055
gemtools, a collection of tools for the downstream and in-depth analysis of structural variants from linked-read data. Gemtools uses the barcoded aligned reads and the Megabase-scale phase blocks to determine haplotypes of structural variant breakpoints and delineate complex breakpoint configurations at the resolution of single DNA molecules.

□ Genetic paradox explained by nonsense:
>> https://www.nature.com/articles/d41586-019-00823-5
upf3a (a member of the nonsense-mediated mRNA decay pathway) and components of the COMPASS complex including wdr5 function in GCR. the GCR is accompanied by an enhancement of histone H3 Lys4 trimethylation (H3K4me3) at the transcription start site regions of the compensatory genes.
□ HLA*LA – HLA typing from linearly projected graph alignments:
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz235/5426702
HLA*LA (“linear alignments”), a graph-based method with high accuracy on exome and low-coverage WGS data, full support for assembled and unassembled long-read data, and a new projection-based approach to graph alignment. HLA*LA improves upon the accuracy of its predecessor HLA*PRG, while being 3-10 times faster and extending HLA typing functionality to long reads and assemblies.

□ Florian BERNARD 🧬
The future is now: cloud-based GPU-enhanced basecalling of @nanopore reads using flipflop. 200K reads in just 30min, for a server cost of ~1$.
It's time to re-basecall EVERYTHING
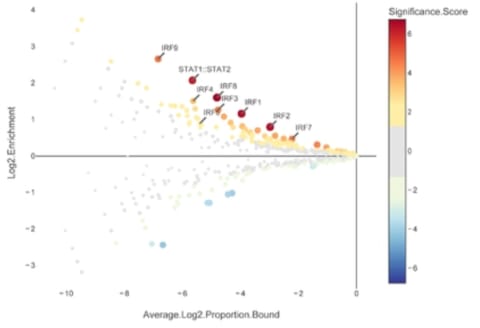
□ CiiiDER: a new tool for predicting and analysing transcription factor binding sites
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/04/599621.full.pdf
CiiiDER performs an enrichment analysis to identify TFs that are significantly over- or under-represented in comparison to a bespoke background set and thereby elucidate pathways regulating sets of genes of pathophysiological importance.

□ Peax: Interactive Visual Pattern Search in Sequential Data Using Unsupervised Deep Representation Learning
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/04/597518.full.pdf
While users label regions as either matching their search target or not, a random forest classifier learns to weigh the importance of different dimensions of the learned representation.

□ Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/04/598748.full.pdf
Vireo (Variational Inference for Reconstructing Ensemble Origins), a principled Bayesian method to demultiplex arbitrary pooled designs that combine genetically distinct individuals.
□ Warped phase coherence: An empirical synchronization measure combining phase and amplitude information
>> https://aip.scitation.org/doi/figure/10.1063/1.5082749
Adding a possibly complex constant value to this normally null-mean signal has a non-trivial warping effect.By means of simulations of Rössler systems and experiments on single-transistor oscillator networks, it is shown that the resulting coherence measure may have an empirical value in improving the inference of the structural couplings from the dynamics.
□ Nano-GLADIATOR: real-time detection of copy number alterations from nanopore sequencing data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz241/5428178










※コメント投稿者のブログIDはブログ作成者のみに通知されます