








 (Photo by Jesse Somera "Affection")
(Photo by Jesse Somera "Affection")
□ A Model of Indel Evolution by Finite-State, Continuous-Time Machines
>> https://www.genetics.org/content/genetics/early/2020/10/06/genetics.120.303630.full.pdf
Seeking to derive the pairwise alignment likelihood directly from an instantaneous model of sequence mutation; that is, a continuous-time Markov chain whose state space is the set of all possible DNA sequences.
The method uses an evolutionary model which can be represented infinitesimally as an HMM can be formally connected to a Pair HMM that approximates its finite-time solution. This may be viewed as an automata-theoretic framing of the Chapman-Kolmogorov equation.
□ Raptor: A fast and space-efficient pre-filter for querying very large collections of nucleotide sequences
>> https://www.biorxiv.org/content/10.1101/2020.10.08.330985v1.full.pdf
Raptor uses winnowing minimizers to define a set of representative k-mers, an extension of the Interleaved Bloom Filters (IBF) as a set membership data structure, and probabilistic thresholding for minimizers.
Raptor uses a set membership data structure, the x-PIBF, to retrieve binning bitvectors. Raptor is ready for secondary memory use and its data structures can be efficiently compressed if the used bitvector is sparse.

□ generative Bayesian Dirichlet-multinomial classifier: Fast and interpretable scRNA-seq data analysis
>> https://www.biorxiv.org/content/10.1101/2020.10.05.314039v1.full.pdf
a Bayesian Dirichlet-multinomial mixture model learns meaningful clusters where the automatically learned relationships between cell types and genes overlap with ground truth associations.
Zero expression renders multinomial based methods are numerically unstable and therefore adding a pseudocount of one to all expression values.
This approach centers on fast Newton-Raphson (NR) optimization for efficiently learning the parameters of a Dirichlet-multinomial (Po ́lya) distribution. the mathematical details of this fast inference protocol as well as how we learn a class-conditional Dir.-mul. model for classification.

□ GraphAligner: rapid and versatile sequence-to-graph alignment
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02157-2
GraphAligner is able to work with a wide range of graphs, including graphs with overlapping as well as non-overlapping node sequences, and accepts GFA as well as vg graph formats.
The bidirected graph is first converted into a directed node-labeled graph which they called the alignment graph. The alignment graph is defined as a directed graph Ga=(Va,Ea⊆(Va×Va),σa=Va→Σn).
GraphAligner, Seed-and-extend strategy for aligning long error-prone reads to genome graphs. GraphAligner uses a bitvector banded DP alignment algorithm to extend the seed hits. The DP matrix is calculated inside a certain area, which depends on the extension parameters.

□ Natrix: A Snakemake-based workflow for processing, clustering, and taxonomically assigning amplicon sequencing reads
>> https://www.biorxiv.org/content/10.1101/2020.09.23.309864v1.full.pdf
Natrix is divided into quality assessment, read assembly, dereplication, chimera detection, split-sample merging, ASV or OTU-generation and taxonomic assessment.
Natrix uses the VSEARCH uchime3 denovo algorithm to detect chimeric sequences. Natrix resolves ASVs without using arbitrary clustering thresholds and with increased resolution. Disjoint paths in the DAG can be executed in parallel.

□ I-CONVEX: Fast and Accurate de Novo Transcriptome Recovery from Long Reads
>> https://www.biorxiv.org/content/10.1101/2020.09.28.317594v1.full.pdf
I-CONVEX is an iterative algorithm for solving "de Novo Transcriptome Recovery from long reads" problem. I-CONVEX performs alignment-free isoform clustering with almost linear computational complexity, and leads to better consensus accuracy on simulated and synthetic datasets.
I-CONVEX does not require read-to-read alignment. I-CONVEX consists of two subprograms: scalable pre-clustering of reads, and alignment-free isoform recovery via convexification. I-CONVEX solves a clustering problem over finite-alphabet sequences.

□ Swan: a library for the analysis and visualization of long-read transcriptomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa836/5912931
Swan works by processing transcript models from either GTF files or from a TALON database into a SwanGraph data structure consisting of a series of data frames and a graph.
Swan provides a platform for deeply exploring full-length transcriptome data. Swan detects novel exon skipping and intron retention events by analyzing the graph models. Transcript novelty categories are determined by TALON.

□ Epiclomal: Probabilistic clustering of sparse single-cell DNA methylation data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008270
Epiclomal, a probabilistic clustering method arising from a hierarchical mixture model to simultaneously cluster sparse scDNA methylation data and impute missing values. Epiclomal can handle the inherent missing data characteristic that dominates single-cell CpG genome sequences.
Epiclomal is a clustering method based on a hierarchical mixture of Bernoulli distributions. Epiclomal uses a principled Variational Bayes inference method that is robust to the initial starting point, with the optimal clustering being obtained multiple times across independent runs.

□ Knockoff Boosted Tree for Model-Free Variable Selection
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa770/5910548
a novel strategy for conducting variable selection without prior model topology knowledge using the knockoff method with boosted tree models.
the sparse covariance and principal component knockoff methods - the PCC knockoff and the sparse Gaussian knockoff. Unlike currently available methods, the PCC knockoff does not depend on Gaussian assumptions for the design matrix.
□ MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02015-1
MOFA+, a model extension addressing the challenges by developing a stochastic variational inference framework amenable to GPU computations, enabling the analysis of datasets with potentially millions of cells and enabling joint modelling of multiple groups and data modalities.
MOFA+ inherits all the features from its predecessor, including a natural approach for handling missing values as well as the capacity to perform inference with non-Gaussian readouts.
□ IsoResolve: Predicting Splice Isoform Functions by Integrating Gene and Isoform-level Features with Domain Adaptation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa829/5910547
IsoResolve is a computational approach for isoform function prediction by leveraging the information of gene function prediction models with domain adaptation. IsoResolve treats gene- level and isoform-level features as source and target domain, respectively.
IsoResolve employs DA to project the two domains to a latent variable - LV space in a way that the LVs projected from the gene and isoform domain features are of the same distribution, enabling that the gene domain information can be leveraged for predicting isoform functions.
□ scDaPars: Dynamic Analysis of Alternative Polyadenylation from Single-Cell RNA-Seq Reveals Cell Subpopulations Invisible to Gene Expression Analysis
>> https://www.biorxiv.org/content/10.1101/2020.09.23.310649v1.full.pdf
scDaPars, a bioinformatics algorithm to accurately quantify APA events at both single-cell and single-gene resolution using standard scRNA-seq data. scDaPars can robustly recover missing APA events caused by the low amounts of mRNA sequenced in single cells.
Since APA exhibits alterations in different cell types and cell states in a global scale, scDaPars recovers missing single-cell level APA dynamics by borrowing information of the same gene from neighboring cells.

□ CarDEC: A Joint Deep Learning Model for Simultaneous Batch Effect Correction, Denoising and Clustering in Single-Cell Transcriptomics
>> https://www.biorxiv.org/content/10.1101/2020.09.23.310003v1.full.pdf
CarDEC (Count a Deep Embedded Clustering), a joint deep learning model that simultaneously clusters and denoises scRNA-seq data, while correcting batch effect both in the embedding and the gene expression space.
CarDEC using a branching architecture that treats highly variablegenes (HVGs) and the remaining genes, which we designate as lowly variable genes (LVGs), as distinct feature blocks.
□ PBSIM2: a simulator for long read sequencers with a novel generative model of quality scores
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa835/5911629
PBSIM2, a generative model for quality scores, in which a hidden Markov Model with a latest model selection method, called Factorized information criteria, is utilized.
In this HMM, the emission probability distributions from each hidden state are provided by a categorical distribution, whose output is one of the quality scores. It should be emphasized that the parameters in categorical distribution with hidden states are different from each other.

□ TALE: Transformer-based protein function Annotation with joint sequence-Label Embedding
>> https://www.biorxiv.org/content/10.1101/2020.09.27.315937v1.full.pdf
TALE replaces previously-used convolutional neural networks (CNN) with self-attention-based transformers which has made a major breakthrough in natural language processing and recently in protein sequence embedding.
Transformers can deal with global dependencies within the sequence in just one layer, which helps detect global sequence patterns for function prediction much easier than CNN. TALE embeds sequence inputs/features and hierarchical function labels (GO terms) into a latent space.

□ Samplot: A Platform for Structural Variant Visual Validation and Automated Filtering
>> https://www.biorxiv.org/content/10.1101/2020.09.23.310110v1.full.pdf
samplot is a command line tool for rapid, multi-sample structural variant visualization. samplot takes SV coordinates and bam files and produces high-quality images that highlight any alignment and depth signals that substantiate the SV.
Samplot allows to focus orthogonal molecular validation assays on smaller groups of variants with far more true-positives. Samplot-ML is a resnet-like model that takes Samplot images of putative deletion SVs as input and predicts a genotype.
□ NoPeak: k-mer based motif discovery in ChIP-Seq data without peak calling
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa845/5912933
NoPeak, a novel approach to reliably identify transcription factor binding motifs from ChIP-Seq data without peak detection.
NoPeak Software the integration profile of k-mers based on mapped reads. Instead of finding peaks across the genome they create read profiles for each k-mer. The profiles have a distinct shape by which they are filtered and scored. Selected k-mers are then combined directly to sequence logos.
□ VFFVA: dynamic load balancing enables large-scale flux variability analysis
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03711-2
The significant contribution is the management of parallelism through a hybrid integration of parallel libraries OpenMP and MPI, for shared memory and non-shared memory systems respectively.
Very Fast Flux Variability Analysis (VFFVA) as a parallel implementation that dynamically balances the computation load between the cores in runtime which guarantees equal convergence time between them.
□ pmTM-align: scalable pairwise and multiple structure alignment with Apache Spark and OpenMP
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03757-2
pmTM-align enables scalable pairwise and multiple structure alignment computing and offers more timely responses for medium to large-sized input data than existing alignment tools such as mTM-align.
pmTM-align employs a hybrid two-stage architecture as Spark can only handle the all-to-all PSA part, while the rest is computed locally with OpenMP support.

□ SC-JNMF: Single-cell clustering integrating multiple quantification methods based on joint non-negative matrix factorization
>> https://www.biorxiv.org/content/10.1101/2020.09.30.319921v1.full.pdf
Matrix factorization is an excellent method for dimension reduction and feature extraction of data. In particular, NMF approximates the data matrix as the product of two matrices in which all factors are non-negative.
SC-JNMF can extract common factors among multiple gene expression profiles by applying each NMF to them under the constraint that one of the factorized matrices is shared among the multiple NMFs.
The coefficient matrix had characteristic factors in each cell cluster, and both basis matrices had similar factors between the matrices. the genes showing high values in the basis matrices probably have important features of the cluster w/ high values in the coefficient matrix.
□ Syncmers are more sensitive than minimizers for selecting conserved k-mers in biological sequences
>> https://www.biorxiv.org/content/10.1101/2020.09.29.319095v1.full.pdf
Unlike a minimizer, a syncmer is identified by its k-mer sequence alone and is therefore synchronized in the following sense: if a given k-mer is selected from one sequence, it will also be selected from any other sequence.
Bounded syncmers are shown to be unambiguously superior to minimizers because they achieve both lower density and better conservation in mutated sequences.
□ A spectral clustering with self-weighted multiple kernel learning method for single-cell RNA-seq data
>> https://academic.oup.com/bib/bib/advance-article-abstract/doi/10.1093/bib/bbaa216/5916937
the performance of a kernel method is largely determined by the selected kernel; a self-weighted multiple kernel learning model can help choose the most suitable kernel for scRNA-seq data.
The main proposition is that automatically learned similarity information from scRNA-seq data is used to transform the candidate solution into a new solution that better approximates the discrete one.
□ BAGS: an automated Barcode, Audit & Grade System for DNA barcode reference libraries
>> https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13262
BAGS performs automated auditing and annotation of cytochrome c oxidase subunit I (COI) sequences libraries, for a given taxonomic group of animals, available in the Barcode of Life Data System (BOLD).
BAGS fulfils a significant gap in the current landscape of DNA barcoding research tools by quickly screening reference libraries to gauge the congruence status of data and facilitate the triage of ambiguous data for posterior review.
□ Adaptive Metropolis-coupled MCMC for BEAST 2
>> https://peerj.com/articles/9473/
an adaptive Metropolis-coupled MCMC scheme to Bayesian phylogenetics, where the temperature difference between heated chains is automatically tuned to achieve a target acceptance probability of states being exchanged between individual chains.
The adaptive MC3 algorithm is compatible with other BEAST 2 packages and therefore works with any implemented model that does not directly affect the MCMC machinery.
□ Testcrosses are an efficient strategy for identifying cis regulatory variation: Bayesian analysis of allele specific expression (BASE)
>> https://www.biorxiv.org/content/10.1101/2020.10.01.322362v1.full.pdf
BASE consists of four main modules: Genotype Specific References, Alignment and SAM Compare, Prior Calculation, and Bayesian Model.
The testcross approach is a useful strategy to maximize allele comparison while minimizing sequencing efforts. Testcrosses will not detect either parent of origin or cis-trans interactions since the comparison between alleles is from a shared maternal/paternal inheritance.

□ A guide to ecosystem models and their environmental applications
>> https://www.nature.com/articles/s41559-020-01298-8
Existing modelling approaches typically attempt to do: describe and disentangle ecosystem components and interactions; make predictions about future ecosystem states; and inform decision making by comparing alternative strategies and identifying important uncertainties.
Ecosystem models that take a dynamical systems theory use a deterministic approach to predict how ecosystems change over time. Such models are typically based on Lotka-Volterra equations, or similar , and have demanding data requirements, especially if the model is complex.

□ Projection in genomic analysis: A theoretical basis to rationalize tensor decomposition and principal component analysis as feature selection tools
>> https://www.biorxiv.org/content/10.1101/2020.10.02.324616v1.full.pdf
explaining why PCA- and TD-based unsupervised FE work well. because singular value vectors correspond to projection onto the centroid subspace obtained by K-means.
empirical threshold adjusted P-values of 0.01 assuming the null hypothesis that singular value vectors attributed to genes obey the Gaussian distribution empirically corresponds to threshold-adjusted P-values of 0.1 when the null distribution is generated by gene order shuffling.

□ Dynamic characteristics rather than static hubs are important in biological networks
>> https://www.biorxiv.org/content/10.1101/2020.09.30.320259v1.full.pdf
a paradigm shift unraveling a new class of nodes different from static hubs and able to determine network dynamics.
This approach is able to depict dynamics without calculating exhaustively the complete network dynamics. Applying it to a variety of biological networks, and identified small sets of nodes sufficient to determine the dynamic behavior of the whole system.

□ KMD clustering: Robust generic clustering of biological data
>> https://www.biorxiv.org/content/10.1101/2020.10.04.325233v1.full.pdf
a generalized silhouette-like function is predictive of clustering accuracy and exploit this property to eliminate the main hyperparameter k. The clustering performance of advanced or specialized clustering algorithms, all of which have cryptic hyperparameters.

□ Baysor: Bayesian segmentation of spatially resolved transcriptomics data
>> https://www.biorxiv.org/content/10.1101/2020.10.05.326777v1.full.pdf
Baysor, a general framework based on Markov Random Fields, that can be used to solve a variety of molecule labeling problems. a Neighbourhood Composition Vector for each molecule by taking its k spatially nearest neighbors and estimating the relative frequency of different genes.

□ Inferring a complete genotype-phenotype map from a small number of measured phenotypes
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008243
The uncertainty inherent in each of the measured and predicted phenotypes will affect the calculated trajectories, but this can be accounted for using a simple sampling strategy to propagate uncertainty in CQ transport to uncertainty in evolutionary trajectories.
The pseudoreplicate genotype-phenotype maps by drawing from the phenotype uncertainties. Because the number of model terms increases linearly, This implementation should be effective even when applied to massive genotype-phenotype maps.
□ H-tSNE: Hierarchical Nonlinear Dimensionality Reduction
>> https://www.biorxiv.org/content/10.1101/2020.10.05.324798v1.full.pdf
Many techniques exist for dimensionality reduction but mainly act as a “black box.” Examples of such methods include Sammon mapping, Curvilinear Components Analysis, SNE, Isomap, Maximum Variance Unfolding, Locally Linear Embedding, and Laplacian Eigenmaps.
H-tSNE formulates a direct relationship between the distance between two graph nodes in the hierarchy and the resulting distance in the embedding.

□ JIND: Joint Integration and Discrimination for Automated Single-Cell Annotation
>> https://www.biorxiv.org/content/10.1101/2020.10.06.327601v1.full.pdf
JIND performs a novel asymmetric alignment in which the transcriptomic profile of unseen cells is mapped onto the previously learned latent space, hence avoiding the need of retraining the model whenever a new dataset becomes available.
The NN used by JIND consists of two subnetworks, an encoder and a classifier. First, the encoder network maps the input gene expression vector onto a 256-dimensional latent space via a one-layer NN.
And refer to the resulting 256-dimensional vector as the latent code, which is then fed into the classifier subnetwork to finally predict the cell-type. These two subnetworks are trained jointly on the source batch by minimizing a weighted categorical cross entropy loss.
□ F-Seq2: improving the feature density based peak caller with dynamic statistics
>> https://www.biorxiv.org/content/10.1101/2020.10.06.328674v1.full.pdf
F-Seq2 combines the power of kernel density estimation and a dynamic “continuous” Poisson distribution to robustly account for local biases and solve ties when ranking candidate peaks.
By combining the power of the local test and the KDE, which model the read probability distribution with statistical rigor, F-Seq2 robustly accounts for local biases and solve ties that occur when ranking candidate summits, making results suitable for IDR analysis.
□ BiSulfite Bolt: A BiSulfite Sequencing Analysis Platform
>> https://www.biorxiv.org/content/10.1101/2020.10.06.328559v1.full.pdf
BiSulfiteBolt (BSBolt), a bisulfite sequencing platform designed to be fast and scalable while also providing the same read-level methylation calls and quality metrics of BS-Seeker2 and Bismark.
BSBolt alignment is built on a forked version BWA-MEM and HTSLIB with bisulfite specific sequencing logic integrated directly into the alignment process. BSBolt includes a rapid and multi-threaded methylation caller.
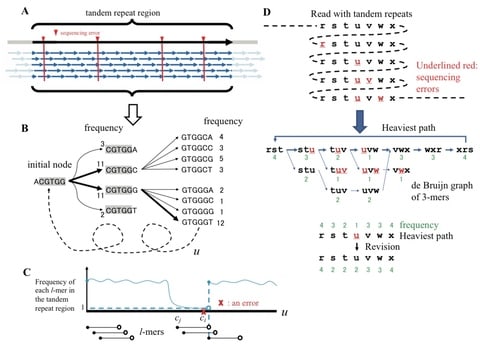
□ Finding Long Tandem Repeats In Long Noisy Reads
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa865/5919583
a long tandem repeat has hundreds or thousands of approximate copies of the repeated unit, so despite the error rate, many short k-mers will be error-free in many copies of the unit.
by analyzing the k-mer frequency distributions of fixed-size windows across the target read, an algorithm that assembles the k-mers of a putative region into the consensus repeat unit by greedily traversing a de Bruijn graph.
This algorithm aligns the representative unit to the input sequence using wraparound dynamic programming and estimates the repeat boundaries.
□ DUBStepR: correlation-based feature selection for clustering single-cell RNA sequencing data
>> https://www.biorxiv.org/content/10.1101/2020.10.07.330563v1.full.pdf
DUBStepR (Determining the Underlying Basis using Stepwise Regression), a feature selection algorithm that leverages gene-gene correlations with a novel measure of inhomogeneity in feature space, termed the Density Index (DI).

□ EMPress enables tree-guided, interactive, and exploratory analyses of multi-omic datasets
>> https://www.biorxiv.org/content/10.1101/2020.10.06.327080v1.full.pdf
By integrating EMPress with the widely-used EMPeror software within QIIME 2, EMPress can simultaneously visualize a phylogenetic tree of features in a study coupled with an ordination of the same study’s samples.










※コメント投稿者のブログIDはブログ作成者のみに通知されます