










□ INTERSTELLAR: A universal sequencing read interpreter
>> https://www.biorxiv.org/content/10.1101/2022.04.16.488535v1.full.pdf
INTERSTELLAR (interpretation, scalable transformation, and emulation of large-scale sequencing reads) that extracts data values encoded in theoretically any type of sequencing read and translates them into sequencing reads of any structure of choice.
INTERSTELLAR enables to translate a more complex read structure with higher order optimal space. A read pool of 10X Chromium multiplexed is translated into a hypothetical structure w/ multi-layered parental-local segment allocations and translated back to the 10X read structure.


□ PanGenie: Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes
>> https://www.nature.com/articles/s41588-022-01043-w
PanGenie, a new algorithm that leverages a haplotype-resolved pangenome reference together with k-mer counts from short-read sequencing data to genotype a wide spectrum of genetic variation - a process is refered to as genome inference.
PanGenie genotypes a large fraction of variants not typable by the former. PanGenie bypasses read mapping and is entirely based on k-mers, which allows it to rapidly proceed from the input short reads to a final callset including SNPs, indels and SVs.
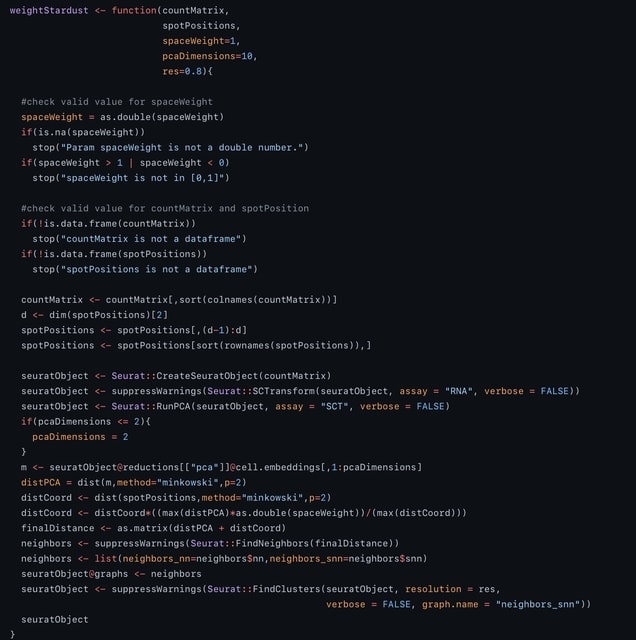
□ Stardust: improving spatial transcriptomics data analysis through space aware modularity optimization based clustering.
>> https://www.biorxiv.org/content/10.1101/2022.04.27.489655v1.full.pdf
spaceWeight defines how much to weigh the space with respect to the transcriptional similarity. By configuring a single parameter the user can control how much the space-based measure weights on the overall measure.
Stardust computes the Louvain edge weights through a linear formulation and requires a fixed a priori parameter. Stardust* uses a dynamic non-linear formulation that changes the spatial weight according to the transcriptomics values in the surrounding space.

□ CellSpace: Scalable sequence-informed embedding of single-cell ATAC-seq data
>> https://www.biorxiv.org/content/10.1101/2022.05.02.490310v1.full.pdf
CellSpace captures meaningful latent structure in scATAC-seq datasets, including cell subpopulations and developmental hierarchies, and scores the activity of transcription factors in single cells based on proximity to binding motifs embedded in the same space.
CellSpace employs a latent embedding algorithm from natural language processing called StarSpace. The latent semantic embedding of entities in StarSpace has also been reformulated as a graph embedding problem.
CellSpace learns a joint embedding of k-mers and cells so that cells will be embedded close to each other in the latent space not simply due to shared accessible events but based on the shared DNA sequence content of their accessible events.

□ Airpart: Interpretable statistical models for analyzing allelic imbalance in single-cell datasets
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac212/6564225
Airpart identifies differential CTS AI from single-cell RNA- sequencing (scRNA-seq) data, or other spatially- or time-resolved datasets. Airpart outputs discrete partitions of data, pointing to groups of genes and cells under common mechanisms of cis-genetic regulation.
Airpart uses a Generalized Fused Lasso with Binomial likelihood for partitioning groups of cells by AI signal, and a hierarchical Bayesian model. Airpart identified DAI patterns across cell states and could be used to define trends of AI signal over spatial or time axes.
<be />

□ scAEGAN: Unification of Single-Cell Genomics Data by Adversarial Learning of Latent Space Correspondences
>> https://www.biorxiv.org/content/10.1101/2022.04.19.488745v1.full.pdf
scAEGAN, a hybrid architecture using an autoencoder (AE) network together with adversarial learning by a cycleGAN (cGAN) network. The core insight is that the AE respects each sample's uniqueness, whereas the cGAN exploits the distributional data similarity in the latent space.
scAEGAN outperforms Seurat3 in library integration, is more robust against data sparsity, and beats Seurat 4 in integrating paired data from the same cell. Furthermore, in predicting one data modality from another, scAEGAN outperforms Babel.

□ GeneVector: Identification of transcriptional programs using dense vector representations defined by mutual information.
>> https://www.biorxiv.org/content/10.1101/2022.04.22.487554v1.full.pdf
GeneVector, a scalable framework for dimensionality reduction implemented as a vector space model using mutual information. It identifies metagenes that correspond to cell-specific transcriptional processes incl. canonical phenotype and cell type-specific interferon activated GE.
GeneVector model provide a framework for identifying metagenes within a gene similarity graph from the cosine distance between each gene vector, and relating these metagenes back to each cell using latent space arithmetic.

□ Poisson VAE: Modeling fragment counts improves single-cell ATAC-seq analysis
>> https://www.biorxiv.org/content/10.1101/2022.05.04.490536v1.full.pdf
scATAC-seq data can be treated quantitatively and that useful information is lost through binarization of the counts. Fragment counts, but not read counts, can be approximately modeled with the Poisson distribution.
Modeling DNA accessibility in single nuclei quantitatively, rather than as a binary state, is consistent with the fact that to access DNA, transcription factors, just like transposases, have to diffuse through the nucleus, likely reaching distinct chromosome territories.
Adapting PeakVI to models Poisson-distributed data in a Poisson VAE. Poisson VAE significantly outperformed PeakVI in reconstructing binarized counts as measured by average precision - NeurIPS: adjusted P = 1.2 x 10^-7 and Satpathy et al.: adjusted P = 6.9 x 10^-8.

□ Clair3-Trio: high-performance Nanopore long-read variant calling in family trios with Trio-to-Trio deep neural networks
>> https://www.biorxiv.org/content/10.1101/2022.05.03.490460v1.full.pdf
The MCVLoss (Mendelian Inheritance Constraint Violation Loss) function is designed to improve variant calling in trios by leveraging the explicit encoding of the priors of the Mendelian inheritance in trios.
Clair3-Trio, the first variant caller tailored for family trio data from Nanopore long-reads. Clair3-Trio employs a Trio-to-Trio deep neural network model, which allows it to input the trio sequencing information and output all of the trio’s predicted variants within a single model.

□ UniTVelo: temporally unified RNA velocity reinforces single-cell trajectory inference
>> https://www.biorxiv.org/content/10.1101/2022.04.27.489808v1.full.pdf
UniTVelo, a statistical framework that models the full dynamics of gene expression with a radial basis function (RBF) and quantifies RNA velocity in a top-down manner. It also introduced a unified latent time across the whole transcriptome.
UniTVelo supports a gene-independent mode to assign the latent time to each gene independently, similar to scVelo. The unified mode allows to aggregate information for all genes, reinforcing the directionality in the trajectory inference, i.e. weak kinetics or complex branches.

□ BiWFA: Optimal gap-affine alignment in O(s) space
>> https://www.biorxiv.org/content/10.1101/2022.04.14.488380v1.full.pdf
the bidirectional Wavefront Alignment algorithm (BiWFA), the first gap-affine algorithm capable of computing optimal alignments in O(s) memory while retaining the WFA’s time complexity of O(ns).
BiWFA’s time complexity is O((m+n)s). BiWFA computes the WFA alignment of two sequences in the forward and reverse direction until they meet. The BiWFA answers the pressing need for sequence alignment methods capable to scaling to genome-scale alignments / full pangenomes.

□ Minigraph-0.17 (r524)
>> https://github.com/lh3/minigraph/releases/tag/v0.17
Minigraph-0.17 gives more accurate graph alignment and generally simpler graph topology. Note that minigraph still focuses on structural variations and does not generate base-level graphs. To endusers, minigraph remains similar feature wise.
Minigraph-0.17 attempts to connect linear chains with the graph wavefront alignemnt algorithm (GWFA) and produces the final alignment with miniwfa under the 2-piece gap penalty. Graph generation also considers base alignment.

□ One Cell At a Time (OCAT): a unified framework to integrate and analyze single-cell RNA-seq data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02659-1
OCAT employs the local anchor embedding (LAE) algorithm to further optimize the edge weights from each single cell to the remaining most similar “ghost” cells, such that the resulting sparsified weights can most effectively reconstruct the transcriptomic features.
OCAT constructs a bipartite graph b/n all single cells and the “ghost” cell set using similarities as edge weights. OCAT captures the cell similarities through message passing b/n the “ghost” cells, which maps the sparsified weights of all single cells to the global latent space.

□ plotsr: Visualising structural similarities and rearrangements between multiple genomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac196/6569079
Plotsr generates high-quality visualisation of synteny and structural rearrangements between multiple genomes. For this, it uses the genomic structural annotations between multiple chromosome-level assemblies.
Plotsr can be used to compare genomes on chromosome level or to zoom in on any selected region. In addition, plotsr can augment the visualisation with regional identifiers (e.g. genes or genomic mark- ers) or histogram tracks for continuous features.

□ UMINT: Unsupervised Neural Network For Single Cell Multi-Omics Integration
>> https://www.biorxiv.org/content/10.1101/2022.04.21.489041v1.full.pdf
UMINT (Unsupervised neural network for single cell Multi-omics INTegration) serves as a promising model for integrating variable number of single cell omics layers with high dimensions, and provides substantial reduction in the number of parameters.
UMINT-generated latent embedding has been proved to produce better clustering as compared to AE. Even without batch integration, UMINT can extract most relevant features from the data that can act as input to further downstream investigations.

□ CONCERT: Genome-wide prediction of sequence elements that modulate DNA replication timing
>> https://www.biorxiv.org/content/10.1101/2022.04.21.488684v1.full.pdf
CONCERT (CONtext-of-sequenCEs for Replication Timing unifies (i) modeling of long-range spatial dependencies across different genomic loci and (ii) detection of a subset of genomic loci that are predictive of the target genomic signals over large-scale spatial domains.
CONCERT integrates two functionally cooperative modules, a selector, which performs importance estimation- based sampling to detect predictive sequence elements, and a predictor, which incorporates bidirectional recurrent neural networks and self-attention mechanism.

□ Generative Moment Matching Networks for Genotype Simulation
>> https://www.biorxiv.org/content/10.1101/2022.04.14.488350v1.full.pdf
Generative Moment Matching Networks (GMMNs) require only training one unique network (the generator), and do not need to observe the data directly, but instead can observe “sketches” that capture the statistical properties of the database as a whole.
GMMN architecture uses a linear layer of dimension 5000 × 4096, followed by a ReLU and a batch norm, followed by another linear layer of dimension 4096 × 5000, finishing w/ a binary quantizer. The random features are implemented w/ a random linear layer of dimension 5000 × 50000.

□ RODAN: a fully convolutional architecture for basecalling nanopore RNA sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04686-y
The RODAN architecture is composed of 22 convolutional blocks and contains around 10M parameters. RODAN gradually incorporates surrounding information for each position in the signal by increasing the kernel size with each successive convolutional block.
In RODAN architecture, increasing the number of channels / the kernel sizes used in each layer, up to 768 channels/a kernel size of 100 in the final layer. the convolutional block includes a pointwise expansion to increase the number of channels before the depthwise convolution.

□ A hybrid unsupervised approach for accurate short read clustering and barcoded sample demultiplexing in nanopore sequencing
>> https://www.biorxiv.org/content/10.1101/2022.04.13.488186v1.full.pdf
An unsupervised hybrid approach to achieve accurate short read clustering for Nanopore sequencing, in which the nucleobase-based greedy algorithm is utilized to obtain initial clusters, and the raw signal information is measured to guide the continuously optimization.
Dynamic Time Warping algorithm has been accelerated by GPU and the clustering time is completely acceptable. A block-wise acceleration strategy is proposed to fully utilize the advantage of GPU blocks, which enables the launch of million threads of DTW calculation simultaneously.

□ deepCNNvalid: Validation of genetic variants from NGS data using Deep Convolutional Neural Networks
>> https://www.biorxiv.org/content/10.1101/2022.04.12.488021v1.full.pdf
The validation of genetic variants can be improved using a machine learning approach resting on a Convolutional Neural Network, trained using ex- isting human annotation. A way in which contextual data from sequencing tracks can be included into the automated assessment.
The idea of including additional context tracks to handle library-specific artefacts translates analogously to this case, so that sequencing data of unrelated samples with the same library preparation would be added along the depth dimension.
□ DiMeLo-seq: a long-read, single-molecule method for mapping protein–DNA interactions genome wide
>> https://www.nature.com/articles/s41592-022-01475-6
DiMeLo-seq combines elements of antibody-directed protein–DNA mapping approache to deposit methylation marks near a specific target protein, then uses long-read sequencing to read out these exogenous methylation marks directly.
DiMeLo-seq’s long sequencing reads often overlap multiple heterozygous sites, enabling phasing and measurement of haplotype-specific protein–DNA interactions. Finally, long reads enable mapping of protein–DNA interactions within highly repetitive regions of the genome.

□ BioNE: Integration of network embeddings for supervised learning
>> https://www.biorxiv.org/content/10.1101/2022.04.26.489560v1.full.pdf
The BioNE framework integrates embeddings from different embedding method, enabling the assessment of whether the combined embeddings offer complementary information with regards to the input network features and thus better performance on prediction tasks.
The BioNE pipeline consists of three steps: network preparation, network embedding, and link prediction: BioNE’s network embedding step takes the prepared input and applies network embedding methods to learn low-dimensional vector representations for each node on the network.

□ METACLUSTERplus - an R package for probabilistic inference and visualization of context-specific transcriptional regulation of biosynthetic gene clusters
>> https://www.biorxiv.org/content/10.1101/2022.04.11.487835v1.full.pdf
METACLUSTERplus, a probabilistic framework that integrates gene expression compendia, context-specific annotations, biosynthetic gene cluster definitions, as well as gene regulatory network architectures.
METACLUSTERplus redefines the transcriptional activity inference in order to compensate for a potential weakness in the original framework. It further augments TA analysis by another layer, that is the simultaneous inference of context specific transcriptional regulation.

□ scTour: a deep learning architecture for robust inference and accurate prediction of cellular dynamics
>> https://www.biorxiv.org/content/10.1101/2022.04.17.488600v1.full.pdf
scTour simultaneously infers the developmental pseudotime, transcriptomic vector field and latent space of cells, with all these inferences unaffected by batch effects inherent in the datasets.
scTour predicts the transcriptomic properties and dynamics of unseen cellular states. the inference of a low-dimensional latent space which combines the intrinsic transcriptome and extrinsic time information provides richer information for reconstructing a finer cell trajectory.

□ PhenoComb: A discovery tool to assess complex phenotypes in high-dimension, single-cell datasets
>> https://www.biorxiv.org/content/10.1101/2022.04.06.487335v1.full.pdf
PhenoComb uses signal intensity thresholds to assign markers to discrete states (e.g. negative, low, high) and then counts the number of cells per sample from all possible marker combinations in a memory-safe manner.
PhenoComb counts the number of cells that have a given phenotype for all possible phenotypes. This is done by first counting cells for all full-length phenotypes, and generating all other phenotypes with neutral states by summing up the cells counted in the full-length ones.

□ Towards a robust out-of-the-box neural network model for genomic data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04660-8
DeepRAM outperforms all other models especially the recurrent version (RNN) in terms of prediction accuracy, overfitting, and robustness across datasets. DeepRAM models are more robust, transferable and generalizable across genomic datasets with varied characteristics.
A LSTM autoencoder model (LSTM-AE) aims to represent a sequence by a dense vector that can be converted back to the original sequence. The encoder reads as input an encoded DNA sequence and outputs a dense vector as the embedding for this sequence whose length is a hyper parameter to tune.
LSTM-AE+NN adds a simple fully connected neural network containing two dense layer with size shrinking by a factor of 2 with a dropout layer in between for the prediction of class labels. The size of the first dense layer is adjusted, as a rule of thumb, to match 1 to 4 times the embedding dimension.
□ DeepCOLOR: Single-cell colocalization analysis using a deep generative model
>> https://www.biorxiv.org/content/10.1101/2022.04.10.487815v1.full.pdf
DeepCOLOR segre- gates cell populations defined by the colocalization relationships and predicts cell-cell interactions between colocalized single cells. DeepCOLOR is typically applicable to studying cell-cell interactions in any spatial niche.
DeepCOLOR was used to build a continuous neural network map from latent cell state space to each spot in the spatial transcriptome in order to enhance consistent mapping profiles between single cells with similar molecular profiles.

□ rox: A statistical model for regression with missing values
>> https://www.biorxiv.org/content/10.1101/2022.04.15.488427v1.full.pdf
rox, “rank order with missing values(X)”, a flexible, non-parametric approach for regression analysis of a dependent variable with missing values and continuous, ordinal, or binary explanatory variables.
rox utilizes the knowledge of missing values representing low concentrations due an Limit Of Detection effect, w/o requiring any actual imputation steps. rox relies on the assumption of an LOD effect in its core, it flexibly generalizes to data with other missingness mechanisms.

□ Karen Miga
>> https://www.nature.com/articles/s41586-022-04601-8
The Human Pangenome Reference Consortium #HPRC aims to create a more complete human reference genome with a graph-based, #T2T representation of global genomic diversity. Exciting perspective from the team released today in @Nature

□ SeATAC: a tool for exploring the chromatin landscape and the role of pioneer factors
>> https://www.biorxiv.org/content/10.1101/2022.04.25.489439v1.full.pdf
SeATAC can be extended to model scATAC-seq data and to investigate the V-plot dynamics. SeATAC uses a conditional variational autoencoder (CVAE) model to learn the latent representation of ATAC-seq V-plots, and to estimate the statistically differential chromatin accessibility.

□ DeepRepeat: direct quantification of short tandem repeats on signal data from nanopore sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02670-6
DeepRepeat accurately detects STRs directly from nanopore electric signals, without using synthetic signals. DeepRepeat is based on the notion that directly adjacent STR units share similar nanopore signal distribution.
DeepRepeat feeds repeat / non-repeat images into aconvolutional neural network followed by a full connection network. Based on alignment of all long reads for a STR locus, the information is summed from multiple long reads for the STR locus using a Gaussian mixture distribution.

□ Oxford Nanopore
>> https://www.nature.com/articles/s41565-022-01116-1
Our own R&D teams complement their work through collaborations with partnerships with academic collaborators. Here, our collaborators at @ucl demonstrating how antibodies can be detected using designed DNA origami nanopores embedded in MinION Flow Cells.

□ GraphPred: An approach to predict multiple DNA motifs from ATAC-seq data using graph neural network and coexisting probability
>> https://www.biorxiv.org/content/10.1101/2022.05.02.490240v1.full.pdf
GraphPred employs a two-layer of GNN. The first layer was used to learn the embedding of k-mer nodes from the similarity graph and coexisting graph, the second layer of GraphPred was used to learn the embedding of sequence nodes from inclusive graph.
GraphPred calculates the coexisting probability of k-mers using the coexisting edges of the heterogeneous graph and finds multiple motifs from an ATAC-seq dataset. GraphPred can capture the important nodes and edges via their weights.

□ PAUSE: Principled Feature Attribution for Unsupervised Gene Expression Analysis
>> https://www.biorxiv.org/content/10.1101/2022.05.03.490535v1.full.pdf
PAUSE, - principled attribution for unsupervised gene expression analysis, combines biologically-constrained autoencoders with principled attributions to improve the unsupervised analysis of gene expression data.
Biologically-constrained “interpretable” autoencoders use prior knowledge to define sparse connections in a deep autoencoder, such that latent variables correspond to the activity of biological pathways.










※コメント投稿者のブログIDはブログ作成者のみに通知されます