□ scEGOT: Single-cell trajectory inference framework based on entropic Gaussian mixture optimal transport
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557102v1
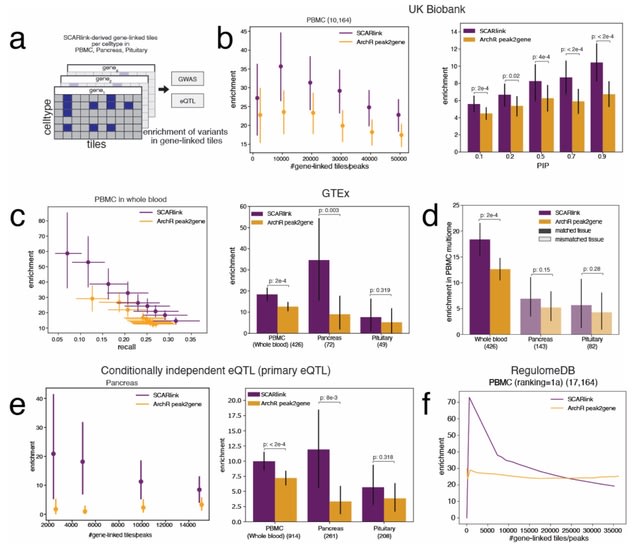
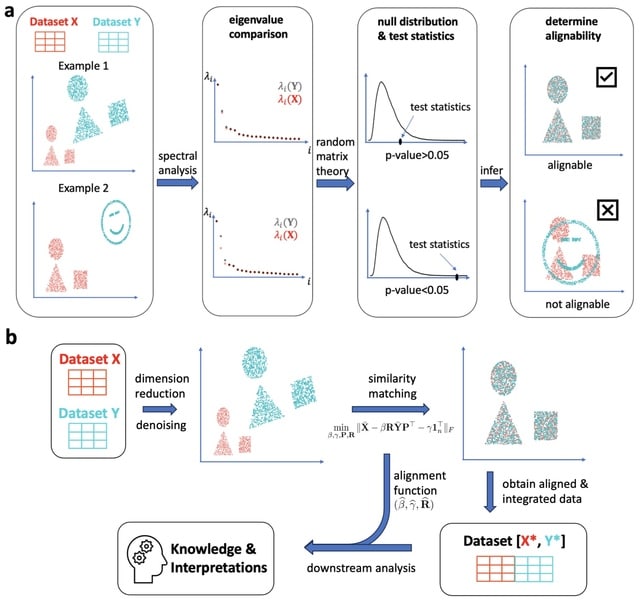
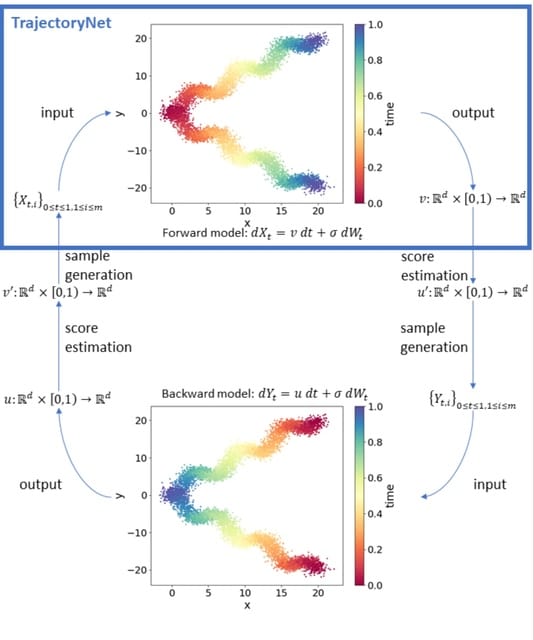
scEGOT provides comprehensive outputs from multiple perspectives, incl. cell state graphs, velocity fields of cell differentiation, time interpolations of single-cell data, space-time continuous GE analysis, GRN, and reconstructions of Waddington’s epigenetic landscape.
scEGOT is formulated by an entropic regularization of the discrete optimal transport, which is a coarse-grained model derived by taking each Gaussian distribution as a single point.
scEGOT constructs the time interpolations of cell populations and the time-continuous gene expression dynamics using the entropic displacement interpolation and has certainly identified the bifurcation time.

□ Cell2Sentence: Teaching Large Language Models the Language of Biology
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557287v1
Cell2Sentence transforms each cell's GE profile into a plaintext of gene names ordered by expression level . This rank transformation can be reverted w/ minimal loss of information. C2S allows any pretrained causal language model (LLMs) to be further fine-tuned on cell sequences.
C2S enables forward / reverse transformation with minimal information loss. Inference is done by generating cells via autoregressive cell completion, generating cells from text, or generating text from cells. The generated cell sentences can be converted back to gene expression.



□ FrameD: Framework for DNA-based Data Storage Design, Verification, and Validation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad572/7274858
FrameD, a software framework for designing, verifying, and validating DNA storage system designs. FrameD is not a library of every conceivable error correction algorithm, instead, it provides a fault-injection-based test bed in which DNA storage systems can be evaluated.
FrameD can be be configured to allocate compute resources in the form of MPI ranks to both fault injection iterations and work done during fault injection simulations like decoding individual strands, packet outer codes, and sequence alignment.

□ DPGA: DNA-based programmable gate arrays for general-purpose DNA computing
>> https://www.nature.com/articles/s41586-023-06484-9
DPGAs, a DIC system by integration of multilayer DNA-based programmable gate arrays. The use of generic single-stranded oligonucleotides as a uniform transmission signal can reliably integrate large-scale DICs with minimal leakage and high fidelity for general-purpose computing.
Reconfiguration of a single DPGA with 24 addressable dual-rail gates can be programmed with wiring instructions to implement over 100 billion distinct circuits.
They designed DNA origami registers to provide the directionality for asynchronous execution of cascaded DPGAs. A quadratic equation-solving DIC assembled with three layers of cascade DPGAs comprising 30 logic gates with around 500 DNA strands.

□ ARES: Geometric deep learning of RNA structure
>> https://www.science.org/doi/10.1126/science.abe5650
The Atomic Rotationally Equivariant Scorer (ARES), predicts the model’s root mean square deviation (RMSD) from the unknown true structure. ARES takes as input a structural model, specified by each atom’s element type and 3D coordinates.
Atom features are repeatedly updated based on the features of nearby atoms. Each feature is then averaged across all atoms, and the resulting averages are fed into additional neural network layers, which output the predicted RMSD of the structural model from the true structure.

□ Allo: Accurate allocation of multi-mapped reads enables regulatory element analysis at repeats
>> https://www.biorxiv.org/content/10.1101/2023.09.12.556916v1
Allo, combines probabilistic mapping based on UMR counts with a convolutional neural network (CNN) that has been trained to identify the appearance of peak-containing regions.
Allo loops through the alignment file and parses uniquely and multi-mapped reads. Alignment files can contain locations that do not have the highest alignment score and thus require extra parsing. Allo identifies the correct pairs when using paired-end sequencing data.
Allo analyzes one read at a time by grouping it with its possible locations. The vector contains the total read count and the output of the sigmoid function. The final score vector is normalized by dividing all entries but the sum of the vector giving the final probabilities.

□ SnapATAC2: a fast, scalable and versatile tool for single-cell omics analysis
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557221v1
SnapATAC2 uses a nonlinear dimensionality reduction algorithm that achieves both computational efficiency and accuracy in discerning cellular composition of complex tissues from a broad spectrum of single-cell omics data types.
SnapATAC2 uses a matrix-free spectral embedding algorithm to project single-cell omics data into a low-dimensional space that preserves the intrinsic geometric properties. SnapATAC2 utilizes the Lanczos algorithm to derive eigenvectors while implicitly using the Laplacian matrix.

□ GraffiTE: a Unified Framework to Analyze Transposable Element Insertion Polymorphisms using Genome-graphs
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557209v1
GraffiTE is a pipeline that finds polymorphic transposable elements (pMEs) in genome assemblies or long read datasets and genotypes the discovered polymorphisms in read sets using a pangenomic approach.
Each pME detected can be further genotyped by mapping short or long reads against a TE graph-genome. It represents each identified ME as a bubble, i.e. providing alternate paths in the graph, where both presence and absence alleles are available for read mapping and genotyping.

□ Cellatlas: Universal preprocessing of single-cell genomics data
>> https://www.biorxiv.org/content/10.1101/2023.09.14.543267v1
Cellatlas is based on parsing of machine-readable seqspec assay specifications to customize inputs for kb-python, which uses kallisto and bustools to catalog reads, error correct barcodes, and count reads.
Cellatlas requires sequencing reads, genomic references, and a seqspec file. It leverages seqspec functionality to auto generate the kallisto string that specifies the 0-index position of the cellular / molecular barcodes, and genomic features such as cDNA or genomic DNA.
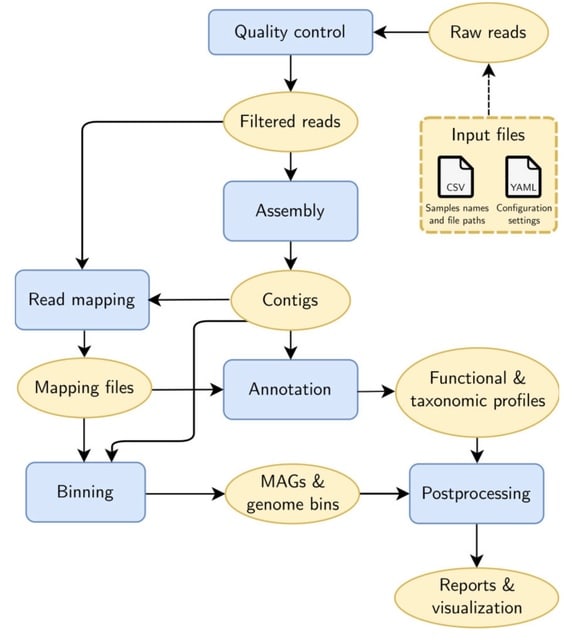
□ Metaphor: A workflow for streamlined assembly and binning of metagenomes https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad055/7233990
Metaphor, a fully automated workflow for genome-resolved metagenomics (GRM). Metaphor differs from existing GRM workflows by offering flexible approaches for the assembly and binning of the input data and by combining multiple binning algorithms with a bin refinement step.
Metaphor produces genome bins generated w/ Vamb / MetaBAT2 / CONCOCT that are refined w/ the DAS Tool. Metaphor processes multiple datasets in a single execution, performing assembly and binning in separate batches for each dataset, and avoiding the need for repeated executions.
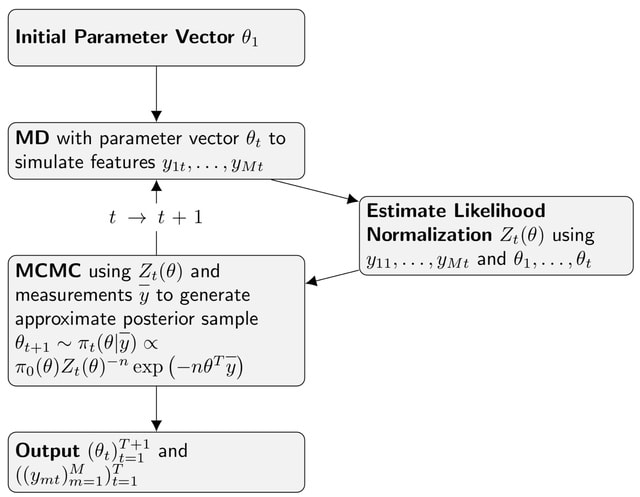
□ Bayesian Maximum Entropy Ensemble Refinement
>> https://www.biorxiv.org/content/10.1101/2023.09.12.557310v1
A fully Bayesian treatment of the estimation of maximum entropy coupling parameters. It tackles the problem head on that the partition function of the maximum entropy ensemble is not tractable analytically.
This approach uses the generated MD trajectories to estimate the partition function using the weighted histogram analysis method (WHAM) algorithm. This achieves an approximation of the maximum entropy Boltzmann probability density, which can be used for MCMC parameter estimation.
This method converges to the maximum entropy ensemble similar to replica averaging, but the limit of infinitely many iterations required in This approach can be systematically improved by simply increasing run time of the algorithm.

□ HILAMA: High-dimensional multi-omic mediation analysis with latent confounding
>> https://www.biorxiv.org/content/10.1101/2023.09.15.557839v1
HILAMA (HIgh-dimensional LAtent-confounding Mediation Analysis) addresses two critical challenges in applying mediation analysis (or any causal inference method) to multi-omics studies: (1) accommodating both high-dimensional exposures and mediators, and (2) handling latent confounding.
Applying HILAMA to a real multi-omic dataset collected by the ADNI. This data analysis should be viewed as at most exploratory rather than confirmatory nature. It is highly likely that the linearity assumption imposed in the Structural Equation Model may not be a good approximation of the reality.

□ UNNT: A novel Utility for comparing Neural Net and Tree-based models
>> https://www.biorxiv.org/content/10.1101/2023.09.12.557300v1
UNNT (A novel Utility for comparing Neural Net and Tree-based models), a novel robust framework that trains and compares deep learning method such as CNN and tree-based method such as XGBoost on the user input dataset.
Grid search trains a new model for ever combination of hvperparameters while cross validation uses a different subset as test data to get an average across five subsets. Best set of hyperparameters found were ETA:0.1, Max depth: 10, Subsample: 0.5, N estimators:500.

□ InterDiff: Guided Diffusion for molecular generation with interaction prompt
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557141v1
InterDiff, an interaction prompt guided diffusion mode. InterDiff is a graph neural network in which the atom denotes the nodes and the Euclidean distance between atoms denotes the edges.
InterDiff consists of 6 equivariant blocks and each block has three modules with transformer like structure. Atoms in ligand and protein are represented by one-hot vector initially and then transformed b a linear laver.

□ MAVEN: compound mechanism of action analysis and visualisation using transcriptomics and compound structure data in R/Shiny
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05416-8
MAVEN (Mechanism of Action Visualisation and Enrichment), an R/Shiny app which allows for GUI-based prediction of drug targets based on chemical structure, combined with causal reasoning based on causal protein–protein interactions and transcriptomic perturbation signatures.
MAVEN is designed to be scalable and flexible to the needs of the user by taking advantage of parallel processing available in PIDGINv4 and CARNIVAL for the two bottleneck steps, and depending on the available resources can handle large networks and gene expression signatures.


□ NAPU: Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation
>> https://www.nature.com/articles/s41592-023-01993-x
Napu (Nanopore Analysis Pipeline) is a collection of WDL workflows for variant calling and de novo assembly of ONT data, optimized for single-flowcell ONT sequencing protocol. A new Hapdup method that generates de novo diploid assemblies from ONT sequencing only.
Outside of centromeres and segmental duplications, These assemblies are structurally highly concordant with the HPRC de novo assemblies that were produced from the more expensive combination of multiple sequencing technologies.

□ EMMA: Computing Multiple Sequence Alignments given a Constraint Subset Alignment
>> https://www.biorxiv.org/content/10.1101/2023.06.12.544642v2
EMMA (Extending Multiple alignments using MAFFT-- add) for the problem of adding a set of unaligned sequences into a multiple sequence alignment (i.e., a constraint alignment).
EMMA builds on MAFFT-- add, which is also designed to add sequences into a given constraint alignment. EMMA improves on MAFFT--add methods by using a divide-and-conquer framework to scale its most accurate version, MAFFT-linsi--add, to constraint alignments with many sequences.


□ Current and future directions in network biology
>> https://arxiv.org/abs/2309.08478
Distinct scientific communities may all analyze biological network data, or they may address identical computational challenges across various application domains, such as biological versus social networks. However, they often do not attend the same research forums.
An algorithmic solution to handling different approach categories is to design hybrid methods that employ techniques from all associated disciplines. Ex. deep learning methods can be combined w/ a network propagation approach to improve the embedding of multiple networks.

□ DeepCAC: a deep learning approach on DNA transcription factors classification based on multi-head self-attention and concatenate convolutional neural network
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05469-9
DeepCAC (Deep Concatenate Attention Augmented Convolution) employs a multi-unit attention mechanism with a convolutional module in the feature extraction layer to form high-dimensional features. DeepCAC can automatically capture heterogeneous hidden features in DNA sequences.
DeepCAC is not designed to apply the Transformer model directly as DNABERT does. The organization of these modules form a complete feature vector by concatenating the feature vector of convolution and the feature vector of multi-head self-attention.

□ General encoding of canonical k-mers
>> https://www.biorxiv.org/content/10.1101/2023.03.09.531845v2
A general minimal perfect hash function for canonical k-mers on alphabets of arbitrary size, i.e., a mapping to the interval [0, σk /2−1]. The approach is introduced for canonicalization under reversal and extended to canonicalization under reverse complementation.
It is formulated recursively where in the i-th step of the recursion, substring x|i, k-i+ 1] is processed. The encoding of a palindromic k-mer solely consists of unspecific pairs until reaching the middle of the k-mer, which is either the empty string or a single character.
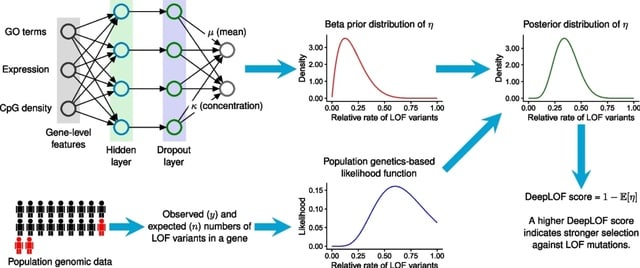
□ DeepLOF: An unsupervised deep learning framework for predicting human essential genes from population and functional genomic data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05481-z
DeepLOF can integrate genomic features and population genomic data to predict LOF-intolerant genes without human-labeled training data. DeepLOF may not suffer from label leakage and other pitfalls of supervised machine learning.
DeepLOF is outperformed by a missense intolerance score, UEECON-G, in the prioritization of dominant-negative disease genes, possibly because many dominant-negative mutations are missense mutations.
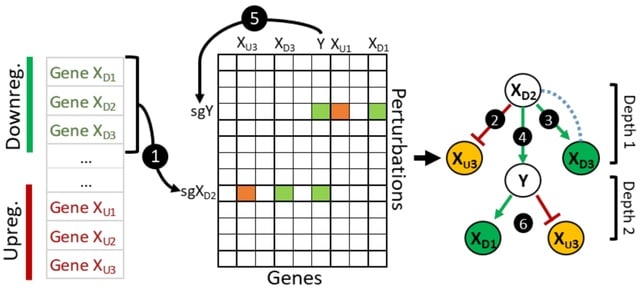
□ GeneSetR: A web server for gene set analysis based on genome-wide Perturb-Seq data
>> https://www.biorxiv.org/content/10.1101/2023.09.18.558211v1
Perturb-Seq based Gene Set Analyzer (GeneSetR), a user-friendly web-server that can analyze user-defined gene lists based on the data from a recently published genome-wide Perturb-Seq study, which targeted 9,866 genes with 11,258 sgRNAs in the K562 cell line.
The GeneSetR encompasses a diverse array of modules, each specifically designed to provide powerful functionalities utilizing the high-dimensional data derived from Perturb-Seq studies.

□ Biastools: easuring, visualizing and diagnosing reference bias
>> https://www.biorxiv.org/content/10.1101/2023.09.13.557552v1
Biastools, a tool for measuring and diagnosing reference bias in datasets from diploid individuals such as humans. biastools enables users to set up and run simulation experiments to compare different alignment programs and reference representations in terms of the bias they yield.
Biastools categorizes instances of reference bias according to their cause, which might be primarily due to genetic differences, repetitiveness, local coordinate ambiguity due to gaps, or other causes.

□ DISCERN: deep single-cell expression reconstruction for improved cell clustering and cell subtype and state detection
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03049-x
DISCERN, a novel deep generative neural network for directed single-cell expression reconstruction. DISCERN allows for the realistic reconstruction of gene expression information by transferring the style of hq data onto lq data, in latent and gene space.
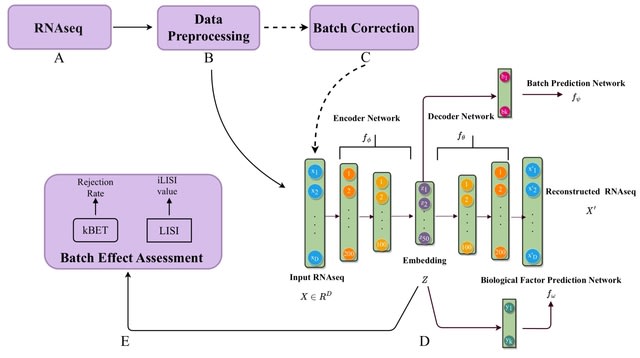
DISCERN is based on a modified Wasserstein Autoencoder. DISCERN transfers the “style” of hq onto lq data to reconstruct missing gene expression, which sets it apart from other batch correction methods such as , which operate in a lower dimensional representation of the data.

□ CLEAN: Targeted decontamination of sequencing data
>> https://www.biorxiv.org/content/10.1101/2023.08.05.552089v2
CLEAN, an easy-to-use all-in-one decontamination pipeline for short reads, long read. CLEAN automatically combines different user-defined FASTA reference sequences, built-in spike-in controls, and downloadable host species into one mapping index for decontamination.
CLEAN concatenates all specified contaminations, e.g., to clean reads of the host and the spike-in in one step. Each input file (FASTQ and/or FASTA) is mapped against the contamination reference with minimap2.

□ meK-Means: Biophysically Interpretable Inference of Cell Types from Multimodal Sequencing Data
>> https://www.biorxiv.org/content/10.1101/2023.09.17.558131v2
meK-Means (mechanistic K-Means), a method to cluster cells from multimodal single-cell data under a self-consistent, biophysical model. Given a set of cell-by-gene count matrices, meK-Means learns clusters of cells which demonstrate shared transcriptional kinetics across genes of interest.
meK-Means infers cluster-specific biophysical parameters which describe transcriptional bursting and rates of mRNA splicing and degradation, alongside learning the partitions of cells into clusters as distinguished by the parameters.

□ Critical assessment of on-premise approaches to scalable genome analysis
> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05470-2
A comprehensive qualitative and quantitative comparison between BCFtools, SnpSift, Hail, GEMINI, and OpenCGA. The tools were compared in terms of data storage technology, query speed, scalability, annotation, data manipulation, visualization, data output representation, and availability.
GEMINI utilizes a Python indexing package called bcolz to speed up queries targeting genotype fields. Genotype columns in the GEMINI database are indexed to accelerate querying using the argument “–use-bcolz” in the same genotype filtering query to get a quick query response.

□ geNomad: Identification of mobile genetic elements https://www.nature.com/articles/s41587-023-01953-y
>> https://www.nature.com/articles/s41587-023-01953-y
geNomad employs a hybrid approach to plasmid and virus identification that combines an alignment-free classifier (sequence branch) and a gene-based classifier (marker branch) to improve classification performance by capitalizing on the strengths of each classifier.
geNomad processes user-provided nucleotide sequences through two branches. In the sequence branch, the inputs are one-hot encoded fed to an IGLOO neural network, which scores inputs based on the detection of non-local sequence motifs.

□ ARA: a flexible pipeline for automated exploration of NCBI SRA datasets
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad067/7243537
The ARA (Automated SRA Records Analysis) tool is implemented in Perl and designed to be used from the shell prompt. It employs the NCBI SRA toolkit to download the raw data in FASTQ format from the SRA database.
ARA provides a full or partial SRA record analysis mode and a choice of the sequence screening method (BLAST and BOWTIE2) and taxonomic profiling (Kraken2). The modular design of the pipeline allows easy further expansion of the sequence analysis toolbox.

□ ANS: Adjusted Neighborhood Scoring to improve assessment of gene signatures in single-cell RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2023.09.20.558114v1
ANS (Adjusted Neighbourhood Scoring) is robust with regard to most influencing factors and returns comparable scores for multiple signatures.
Although all scoring methods demonstrate resilience against variations in data composition and variability in signature qualities, they do not exhibit, except for ANS, comparable score ranges for gene signatures designed to discriminate similar cell types.
 □ Low-input and single-cell methods for Infinium DNA methylation BeadChips
□ Low-input and single-cell methods for Infinium DNA methylation BeadChips >> https://www.biorxiv.org/content/10.1101/2023.09.18.558252v1
A new signal detection framework to address the computational challenge of processing data from limited DNA. This new method significantly improved array detection rates while effectively masking probes whose readings are dominated by background signals.
The Infinium BeadChip is compatible with samples of low input down to single cells. The modified detection p-values calculation achieved higher sensitivities for low-input datasets and was validated in over 100,000 public datasets with diverse methylation profiles.

□ MANOCCA: A multivariate outcome test of covariance
>> https://www.biorxiv.org/content/10.1101/2023.09.20.558234v1
MANOCCA (Multivariate Analysis of Conditional CovAriance) enables the identification of both categorical and continuous predictors associated with changes in the covariance matrix of a multivariate outcome while allowing for covariates adjustment.
MANOCCA outperforms existing covariance methods and that, given the appropriate parametrization, it can maintain a calibrated type I error in a range of realistic scenarios when analysing highly multidimensional data.

□ Fast and sensitive validation of fusion transcripts in whole-genome sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05489-5
A pipeline to validate gene fusions found in RNA-Seq data at the WGS level. The pipeline consists of extracting, processing and filtering discordant read pairs from specific areas of the genome defined by the detected fusion junctions of fusion transcripts.
The regions to search for discordant read pairs are defined by the junction coordinates of the observed fusion transcript.
Genomic evidence for a fusion will theoretically be found downstream of the sequences observed on fusion transcript for the 5′ partner and upstream for the 3′ partner, thereby limiting the region needed to search for discordant reads.












































































































 (Art by
(Art by 





























































 (Art taken from the Terrence Malicks film “
(Art taken from the Terrence Malicks film “






























 (Generative Art by
(Generative Art by