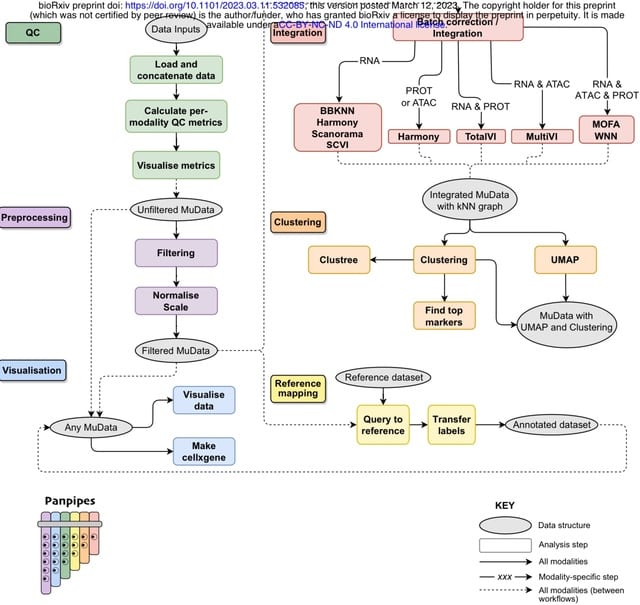
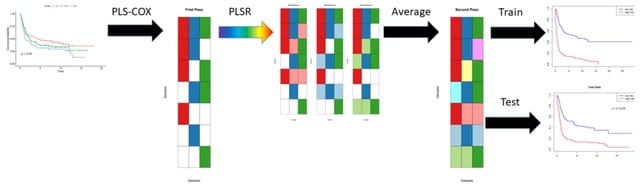
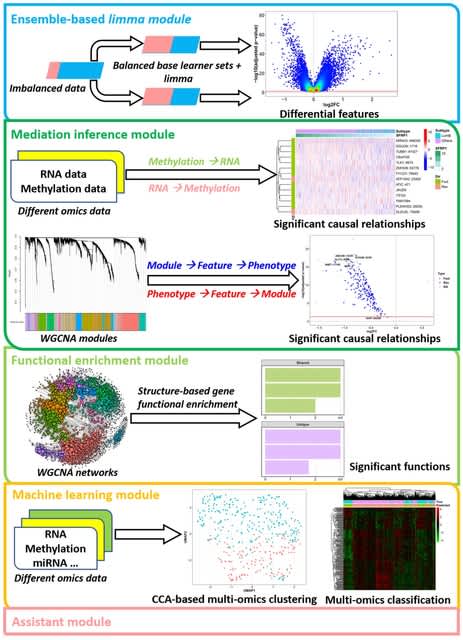
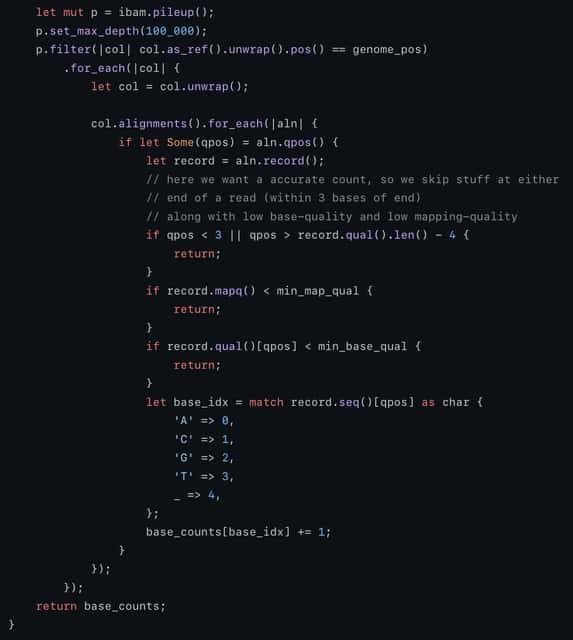
□ scANNA: Boosting Single-Cell RNA Sequencing Analysis with Simple Neural Attention
>> https://www.biorxiv.org/content/10.1101/2023.05.29.542760v1
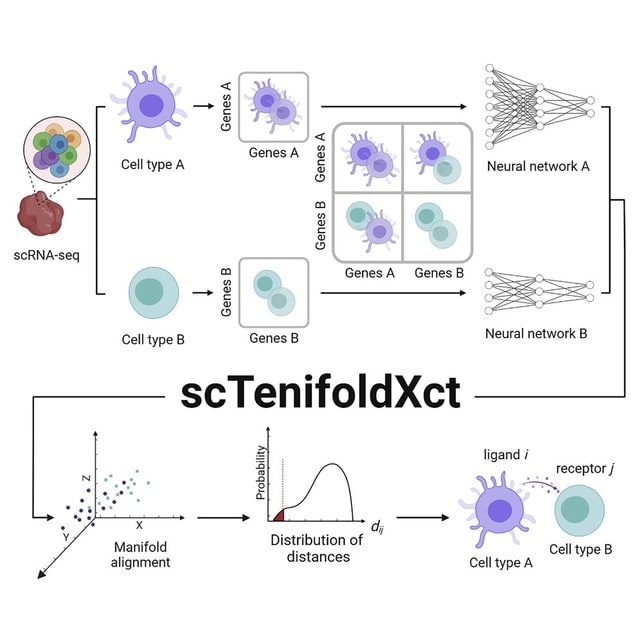
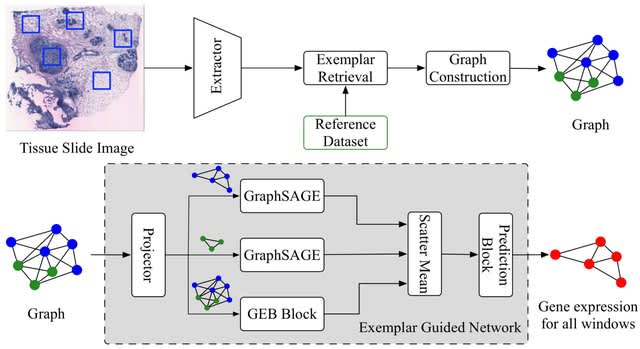
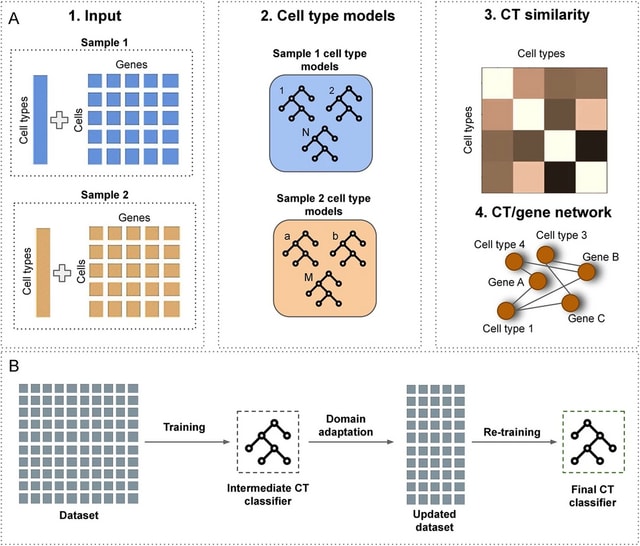
scANNA (single-cell Analysis using Neural-Attention) learns salient genes for each cluster enabling accurate / scalable unsupervised annotations. After training scANNA's DL core, the gene attention weights from the Additive Attention Module are used as input for downstream tasks.
scANNA uses the Deep Projection Blocks, which are an ensemble of operators learning a nonlinear mapping between gene scores. This mapping is designed
to increase model capacity and connect the gene associations to the auxiliary objective.

□ COMSE: Analysis of Single-Cell RNA-seq Data Using Community Detection Based Feature Selection
>> https://www.biorxiv.org/content/10.1101/2023.06.03.543526v1
COMSE partitions all genes into different communities in latent space using the Louvain algorithm. A denoising procedure removes noise introduced during sequencing or other procedures. It then selects highly informative genes from each community based on the Laplacian score.
COMSE calculates the Laplacian score with multi-subsample randomization and choose genes with the smallest scores, assuming that data from the same class are often close to each other. COMSE then rank the genes based on gene-gene correlation to remove redundancy.

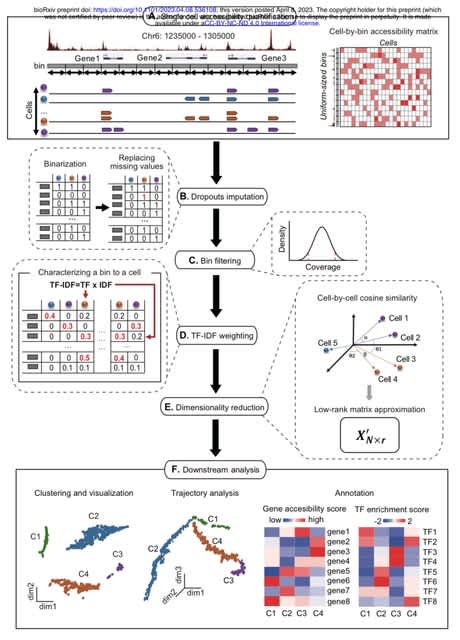
□ scATAnno: Automated Cell Type Annotation for single-cell ATAC-seq Data
>> https://www.biorxiv.org/content/10.1101/2023.06.01.543296v1
scATAnno, a workflow that directly and automatically annotates scATAC-seq data based on scATAC-seq reference atlases. scATAnno directly uses peaks or CRE genomic regions as input features, eliminating the need to convert the epigenomic features into gene activity scores.
scATAnno uses chromatin state profile of large-scale reference atlas to generate peak signals and reference peaks. scATAnno tackles the high dimensionality of SCATAC-seq data by leveraging spectral embedding to efficiently transform the data into a low dimensional space.
Each query cell is assigned a cell type along with two uncertainty scores: the first uncertainty score is based on the KNN, and the second uncertainty score is derived from a novel computation of the weighted distance between the query cell and reference cell type centroids.

□ Sophia: A Scalable Stochastic Second-order Optimizer for Language Model Pre-training
>> https://arxiv.org/abs/2305.14342
Sophia, Second-order Clipped Stochastic Optimization, a simple scalable second-order optimizer that uses a light-weight estimate of the diagonal Hessian as the pre-conditioner. Sophia only estimates the diagonal Hessian every handful of iterations.
The update is the moving average of the gradients divided by the moving average of the estimated Hessian, followed by element-wise clipping. It controls the worst-case update size and tames the negative impact of non-convexity and rapid change of Hessian along the trajectory.
Sophia has a more aggressive pre-conditioner than Adam Sophia applies a stronger penalization to updates in sharp dimensions (where the Hessian is large) than the flat dimensions (where the Hessian is small), ensuring a uniform loss decrease across all parameter dimensions.

□ AlphaDev: Faster sorting algorithms discovered using deep reinforcement learning
>> https://www.nature.com/articles/s41586-023-06004-9
Formulating the problem of discovering new, efficient sorting algorithms as a single-player game that they refer to as AssemblyGame. The AlphaDev learning algorithm can incorporate both DRL as well as stochastic search optimization algorithms to play AssemblyGame.
The primary AlphaDev representation is based on Transformers. AlphaDev discovered small sorting algorithms from scratch that outperformed previously known human benchmarks. These algorithms have been integrated into the LLVM standard C++ sort library.

□ NOS: diffusioN Optimized Sampling: Protein Design with Guided Discrete Diffusion
>> https://arxiv.org/abs/2305.20009
NOS, a guidance method for discrete diffusion models that follows gradients in the hidden states of the denoising network. NOS can perform design directly in sequence space, circumventing significant limitations of structure-based methods, incl. scarce data and inverse design.
NOS generalizes LaMBO, a Bayesian optimization procedure for sequence design that facilitates multiple objectives and edit-based constraints. The resulting method, LaMBO-2, enables discrete diffusions and stronger performance through a novel application of saliency maps.

□ MISATO - Machine learning dataset for structure-based drug discovery
>> https://www.biorxiv.org/content/10.1101/2023.05.24.542082v1
MISATO, a curated dataset of 20000 experimental structures of protein-ligand complexes, associated molecular dynamics traces, and electronic properties. Semi-empirical quantum mechanics was used to systematically refine protonation states of proteins and small molecule ligands.
Molecular dynamics traces for protein-ligand complexes were obtained in explicit water. The dataset is made readily available to the scientific community via simple python data-loaders. AI baseline models are provided for dynamical and electronic properties.

□ SifiNet: A robust and accurate method to identify feature gene sets and annotate cells
>> https://www.biorxiv.org/content/10.1101/2023.05.24.541352v1
SifiNet (Single-cell feature identification w/ Network topology), a cell-clustering-independent method for directly identifying feature gene sets. SifiNet is based on the observation that co-differentially-expressed genes w/ a cell subpopulation exhibit co-expression patterns.
SifiNet constructs a gene co-expression network and explores its topology to identify feature gene sets. It also applies to scATAC-seq data, generating a gene co-open-chromatin network and exploring network topology to identify epigenomic feature gene sets.

□ scTIE: data integration and inference of gene regulation using single-cell temporal multimodal data
>> https://www.biorxiv.org/content/10.1101/2023.05.18.541381v1
scTIE, an autoencoder-based method for integrating multimodal profiling of scRNA-seq / scATAC-seq data over a time course. scTIE provides the first unified framework for the integration of temporal data and the inference of context-specific GRNs that predict cell fates.
scTIE uses iterative optimal transport (OT) fitting to align cells in similar states between different time points and estimate their transition probabilities. scTIE removes the need for selecting highly variable genes (HVGs) as input through a pair of coupled batchnorm layers.
scTIE provides the means to extract interpretable features from the embedding space by linking the developmental trajectories of cell representations. scTIE formulates a trajectory prediction using the estimated transition probabilities and uses gradient-based saliency mapping.

□ scME: A Dual-Modality Factor Model for Single-Cell Multi-Omics Embedding https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad337/7176368
scME can generate a better joint representation of multiple modalities than those generated by other single-cell multi-omics integration algorithms, which gives a clear elucidation of nuanced differences among cells.
scME relies on clustering to determine the shared and complementary information between modalities. Hence, the parameters of a clustering algorithm, such as resolution of the Leiden algorithm, could affect the efficacy of this algorithm.

□ scBalance – a scalable sparse neural network framework for rare cell type annotation of single-cell transcriptome data
>> https://www.nature.com/articles/s42003-023-04928-6
scBalance, a sparse neural network framework that can automatically label rare cell types in scRNA-seq datasets of all scales. scBalance will automatically choose the weight for each cell type in the reference dataset and construct the training batch.
scBalance leverages the combination of weight sampling and sparse neural network, whereby minor (rare) cell types are more informative without harming the annotation efficiency of the common (major) cell populations.
scBalance will iteratively learn mini batches from a three-layer neural network until the cross-entropy loss converges. In the training stage, scBalance randomly disables neurons in the network.

□ SIMBA: single-cell embedding along with features
>> https://www.nature.com/articles/s41592-023-01899-8
SIMBA is a single-cell embedding method that supports single- or multi-modality analyses. It leverages recent graph embedding techniques to embed cells and genomic features into a shared latent space.
SIMBA introduces several crucial procedures, including Softmax transformation, weight decay for controlling overfitting and entity-type constraints to generate comparable embeddings (co-embeddings) of cells and features and to address unique challenges in single-cell data.

□ gRNAde: Multi-State RNA Design with Geometric Multi-Graph Neural Networks
>> https://arxiv.org/abs/2305.14749
gRNAde, a geometric deep learning-based pipeline for RNA sequence design conditioned on multiple backbone conformations.
gRNAde explicitly accounts for RNA conformational flexibility via a novel multi-Graph Neural Network architecture which independently encodes a set of conformers via message passing, followed by conformer order-invariant pooling and sequence design.

□ Cellenium—a scalable and interactive visual analytics app for exploring multimodal single-cell data
>>
Cellenium, a full-stack scalable visual analytics web application which enables users to semantically integrate and organize all their single-cell RNA-, ATAC- , and CITE-sequencing studies.
Cellenium consists of a central Postgres database for hosting all expression- and meta-data, a Postgraphile based GraphQL API layer. Cellenium precalculates differential gene expressions between each annotated cell type and all other cells.

□ Lineage motifs: developmental modules for control of cell type proportions
>> https://www.biorxiv.org/content/10.1101/2023.06.06.543925v1
Lineage Motif Analysis (LMA), a method that recursively identifies statistically overrepresented patterns of cell fates on lineage trees as potential signatures of committed progenitor states.
LMA is based on motif detection, which has been used to identify the building blocks of complex regulatory networks, DNA sequences, and other biological features.
Motifs could be generated by progenitors intrinsically programmed to autonomously give rise to specific patterns of descendant cell fates. It reflects developmental programs invl. extrinsic cues and cell-cell signaling that generate correlated cell fate patterns on lineage trees.

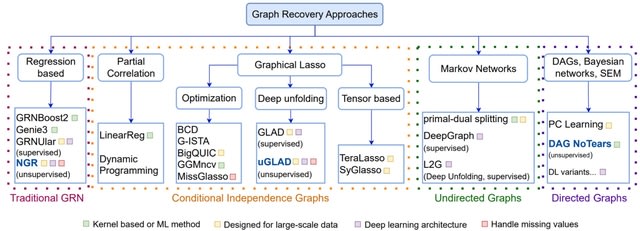
□ MCPNet: A parallel maximum capacity-based genome-scale gene network construction framework
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad373/7192172
MCP (Maximum Capacity Path) Score, a novel maximum-capacity-path based metric to quantify the relative strengths of direct and indirect gene-gene interactions. MCPNet combines interactions from multiple path lengths using optimized weights identified with partial groundtruth.

□ Spider: a flexible and unified framework for simulating spatial transcriptomics data
>> https://www.biorxiv.org/content/10.1101/2023.05.21.541605v1
Spider generates locations of cells on a plate randomly or in a uniform grid-like pattern. Spider supports various neighborhood metrics, such as k-nearest neighbors or neighbors identified by Delaunay triangulation.

□ SanntiS: Expansion of novel biosynthetic gene clusters from diverse environments
>> https://www.biorxiv.org/content/10.1101/2023.05.23.540769v1
At the core of SanntiS is the detection model, an Artificial Neural Network with a one-dimensional convolutional layer, plus a BiLSTM. The model was developed using linearized sequences of protein annotations based on a subset of InterPro as input.
SanntiS employes a duration robust loss function (RLF). RLF mitigates the issue of class imbalance, which can arise from the disparities in BGC counts by class and the variation in the duration of detection events - the disparities in length across different BGC classes.

□ Identification of Biochemical Pathways Responsible for Distinct Phenotypes Using Gene Ontology Causal Activity Models
>> https://www.biorxiv.org/content/10.1101/2023.05.22.541760v1
Phenotypic variability among affected individuals described as incomplete penetrance and variable expressivity can be the result of interactions between the mutated gene and other genes with which it normally interacts.
Integrating the information about human biology from Reactome with model-organism biology from MGI. It can be used not only to understand the similarities of the pathways but as a testing ground for manipulation of pathways in more experimentally tractable organisms than human.

□ MetaBayesDTA: codeless Bayesian meta-analysis of test accuracy, with or without a gold standard
>> https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-023-01910-y
MetaBayesDTA uses the bivariate model to conduct analysis assuming a perfect reference test, and users can also conduct univariate meta-regression and subgroup analysis. It uses latent class models (LCMs) to conduct analyses without assuming a perfect gold standard.
MetaBayesDTA allows the user to run models assuming conditional independence or dependence, options for whether to model the reference and index test sensitivities and specificities as fixed or random effects, and can model multiple reference tests using a meta-regression covariate.

□ WebAtlas pipeline for integrated single cell and spatial transcriptomic data
>> https://www.biorxiv.org/content/10.1101/2023.05.19.541329v1
WebAtlas incorporates integrated scRNA-seq, imaging- and sequencing-based ST datasets for interactive web visualisation, enabling cross-query of cell types and gene expressions across modalities.
WebAtlas unifies commonly used atlassing technologies into the cloud-optimised Zarr format and builds on Vitessce to enable remote data navigation. On WebAtlas, single cell and spatial datasets are linked by biomolecular metadata.
Linkage is performed prior to WebAtlas ingestion using existing data integration methods like Cell2location and StabMap that map scRNA-seq cell type references onto ST datasets and impute unobserved gene expression in the latter.

□ ROCCO: A Robust Method for Detection of Open Chromatin via Convex Optimization
>> https://www.biorxiv.org/content/10.1101/2023.05.24.542132v1
ROCCO determines consensus open chromatin regions across multiple samples simultaneously. ROCCO uses robust summary statistics across samples by solving a constrained optimization problem formulated to account for both enrichment & spatial features of open chromatin signal data.
The model accounts for features common to the edges of accessible chromatin regions, which are often hard to determine based on independently determined sample peaks that can vary widely in their genomic locations.

□ FuzzyPPI: Human Proteome at Fuzzy Semantic Space
>> https://www.biorxiv.org/content/10.1101/2023.05.24.541959v1
FuzzyPPI, a fuzzy semantic scoring function using the Gene Ontology (GO) graphs to assess the binding affinity between any two proteins at an organism level.
FuzzyPPI also constructs a fuzzy semantic network at proteome level from the above designed binding affinity function and extraction of meaningful biological insights.

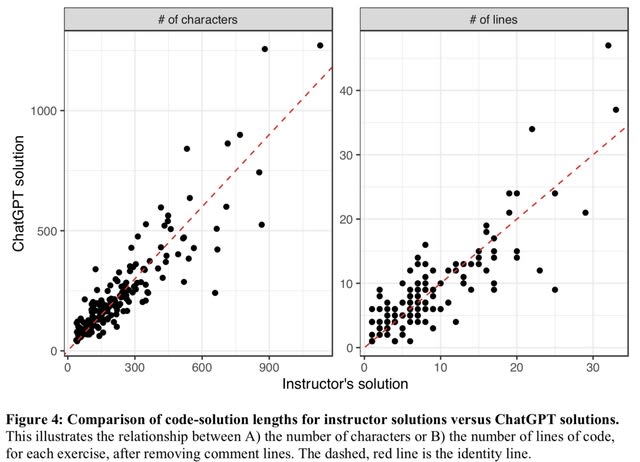
□ Classifying high-dimensional phenotypes with ensemble learning
>> https://www.biorxiv.org/content/10.1101/2023.05.29.542750v1
A meta-analysis of 33 algorithms across 20 datasets containing over 20,000 high-dimensional shape phenotypes using an ensemble learning framework. Both binary and multi-class (e.g., species, genotype, population) classification tasks were considered.
They employs phenotypic datasets containing a range of anatomical data from different organisms with unique class distributions. Blending ensemble approaches involve strategically stacking a set of individual classifiers using a holdout validation set to improve performance.

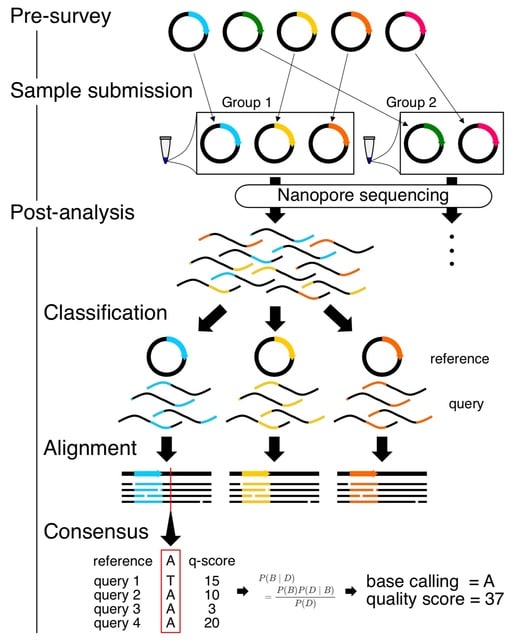
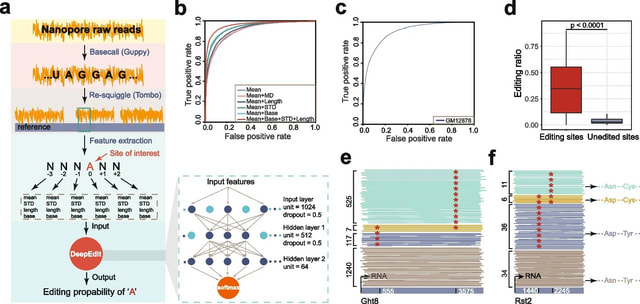
□ buttery-eel: Accelerated nanopore basecalling with SLOW5 data format
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad352/7186500
Buttery-eel, an open-source wrapper for Oxford Nanopore’s Guppy basecaller that enables SLOW5 data access, resulting in performance improvements that are essential for scalable, affordable basecalling.
Buttery-eel/BLOW5 demonstrates a ~3-fold performance improvement when using FAST basecalling, compared to ~20% improvement with HAC basecalling. This suggests that there is an underlying bottleneck in data access on the PromethION.

□ FAST: Flexible Analysis of Spatial Transcriptomics Data: A Deconvolution Approach
>> https://www.biorxiv.org/content/10.1101/2023.05.26.542550v1
A novel reference-free method based on regularized non-negative matrix factorization (NMF), named Flexible Analysis of Spatial Transcriptomics (FAST), that can effectively incorporate gene expression data, spatial coordinates, and histology information into a unified deconvolution framework.
FADT is adaptable to any graph Laplacian matrix, allowing for flexibility in its application. The second term imposes a constraint on cell proportions, encouraging their summation equals one.

□ autoStreamTree: Genomic variant data fitted to geospatial networks
>> https://www.biorxiv.org/content/10.1101/2023.05.27.542562v1
autoStreamTree provides a companion library of functions for calculating various measures of genetic distances among individuals or populations, including model-corrected p-distances as well as those based on allele frequencies.
autoStreamTree includes integrated functions for parsing an input vector shapefile of streams for calculation of pairwise stream distances b/n sites, as well as the ordinary or weighted least-squares fitting of reach-wise genetic distances according to the "stream tree" model.

□ Hierarchical Interleaved Bloom Filter: enabling ultrafast, approximate sequence queries
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02971-4
the Hierarchical Interleaved Bloom Filter (HIBF) that overcomes major limitations of the IBF data structure. The HIBF successfully decouples the user input from the internal representation, enabling it to handle unbalanced size distributions and millions of samples.
The HIBF structure has enormous potential. It can be used on its own, like in the tool Raptor, or can serve as a prefilter to distribute more advanced analyses such as read mapping. Querying ten million reads could be done by querying 11 HIBFs on different machines in parallel.

□ A survey of mapping algorithms in the long-reads era
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02972-3
Adapting and tailoring long-read aligners to such applications will significantly improve analysis over the limited possibilities existing with short reads. Moreover, using pangenomes represented as graphs made from a set of reference genomes is becoming more prevalent.
As a result, long-read mapping to these structures is a novel and active field for genomic reads but should soon expand to other applications such as transcriptomics.
Notably, pangenome graphs vary in definition and structure (overlap graphs, de Bruijn graphs, graphs of minimizers) and therefore expect a diversified algorithmic response to mapping sequences on these graphs.

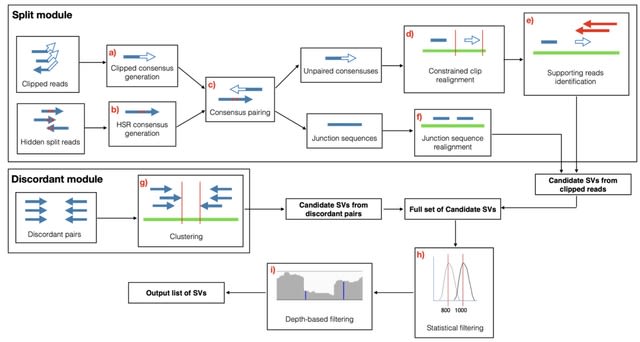
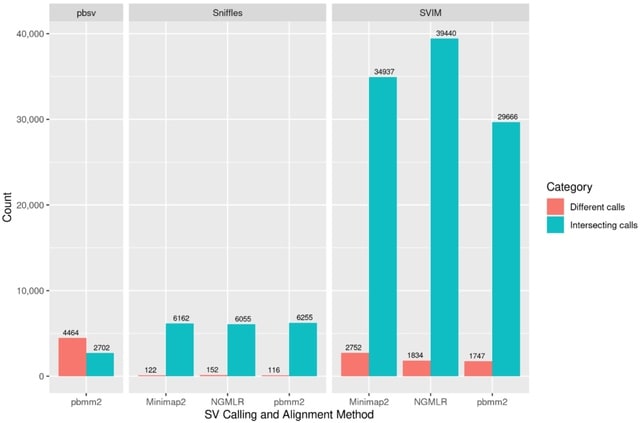
□ SVcnn: an accurate deep learning-based method for detecting structural variation based on long-read data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05324-x
SVcnn accurately detects DELs, INSs, DUPs, and INVs. SVcnn is a convolutional neural network (CNN) based method. It uses hierarchical clustering to identify if a region contains multi-allelic SVs. Moreover, SVcnn utilizes the LetNet model to distinguish whether an SV is a true SV or not.
The input of SVcnn consists of (i) a sorted long read bam file and (ii) a reference file. SVcnn mainly consists of three main steps: (1) Detecting candidate SVs, (2) Converting to image and building model, (3) Filtering and outputting SVs.

□ Epiphany: predicting Hi-C contact maps from 1D epigenomic signals
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02934-9
Epiphany, a neural network to predict cell-type-specific Hi-C contact maps from widely available epigenomic tracks. Epiphany uses Bi-LSTM layers to capture long-range dependencies and optionally a generative adversarial network architecture to encourage contact map realism.
Epiphany can be trained with MSE alone or with a combination of MSE and GAN loss. In the latter case, the full model consists of two parts: a generator to extract information and make predictions, and a discriminator to introduce adversarial loss into the training process.

□ networkGWAS: A network-based approach to discover genetic associations
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad370/7191773
networkGWAS, a statistically sound approach to network-based GWAS using mixed models and neighborhood aggregation. It allows for population structure correction and for well-calibrated p-values, which are obtained through circular and degree-preserving network permutations.
networkGWAS successfully detects known associations on diverse synthetic phenotypes. It employs a FaST-LMM-Set like model to estimate the statistical associations with the phenotype of choice. networkGWAS presents higher recall in comparison to dmGWAS per each precision value.

□ NoVaTeST: Identifying Genes with Location Dependent Noise Variance in Spatial Transcriptomics Data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad372/7191774
NoVaTeST pipeline that offers a more general spatial gene expression modeling in ST data using the heteroscedastic Gaussian process. The pipeline uses Wilcoxon signed rank test and FDR correction to identify genes with location-dependent noise variance.

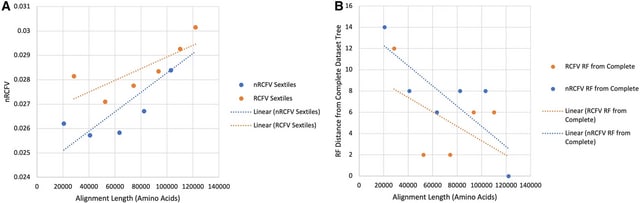
□ TreeTerminus - Creating transcript trees using inferential replicate counts
>> https://www.sciencedirect.com/science/article/pii/S2589004223010386
TreeTerminus, a data-driven approach for grouping transcripts into a tree structure where leaves represent individual transcripts and internal nodes represent an aggregation of a transcript set.
TreeTerminus constructs trees such that, on average, the inferential uncertainty decreases as ascending the tree topology. It provides the flexibility to analyze data at nodes that are at different levels of resolution and can be tuned depending on the analysis of interest.


































 (Art by
(Art by 


























































 (Art by
(Art by 














































 (Art by Beau Wright)
(Art by Beau Wright)