











□ BertNDA: a Model Based on Graph-Bert and Multi-scale Information Fusion for ncRNA-disease Association Prediction
>> https://www.biorxiv.org/content/10.1101/2023.05.18.541387v1
BertNDA employs Laplace transform of graph structure and WL(Weisfeiler-Lehman) absolute role coding to extract global information. Construct a connectionless subgraph to aggregate neighbor feature to identify local information.
An EMLP (Element-weight MLP) structure is adopted to obtain the multi-scale feature representation of node. Furtherly, nodes are encoded using Transformer-encoder structure. BertNDA acquires the semantic similarity and Gaussian interaction profile kernel similarity matrix.
BertNDA calculates the Laplace matrix on the structure of the entire graph after data preprocessing. Eigenvectors are defined via the factorization of the graph Laplacian matrix. The absolute role embedding of nodes is calculated by using the WL algorithm.

□ Geneformer: Transfer learning enables predictions in network biology
>> https://www.nature.com/articles/s41586-023-06139-9
Geneformer, a context-aware, attention-based deep learning model, pretrained on a large-scale corpus of about 30 million single-cell transcriptomes to enable context-specific predictions in settings with limited data in network biology.
Geneformer encodes network hierarchy in the attention weights of the model in a completely self-supervised manner. Fine-tuning towards a diverse panel of downstream tasks relevant to chromatin and network dynamics. Geneformer consistently boosted predictive accuracy.

□ Dimension reduction of dynamics on modular and heterogeneous directed networks
>> https://academic.oup.com/pnasnexus/article/2/5/pgad150/7147610
A method for reducing a given N-dimensional dynamical system on a network into an n-dimensional one whose variables, the observables, represent weighted averages of the node activities A reduced adjacency matrix and an approximate system of ODEs for the observables’ evolution.
Calculating the reduction vectors that are used to construct the observables from the node activities. These vectors fully determine the reduced approximate dynamics, incl. a reduced adjacency matrix that specifies the magnitude of the coupling between observables.

□ xRead: a coverage-guided approach for scalable construction of read overlapping graph
>> https://www.biorxiv.org/content/10.1101/2023.05.23.541864v1
×Read keeps a global graph data structure to record read overlaps during the iterative process. The produced alignment skeletons are converted to read overlapping information and supplied to the data structure incrementally.
For a given query read, the produced alignment skeletons that meet one of the following three conditions are filtered out at first since they could be false positives caused by sequencing errors or repeats in local genomic regions:
×Read (re-)estimates read coverages w/ the updated overlapping information. For a given read, its coverage is estimated by the numbers of the seed reads directly connected to it by the CROs. The reads having CROs to the same seed reads which can be regarded as indirectly aligned.

□ NS-DIMCORN: Ordinary differential equations to construct invertible generative models of cell type and tissue-specific regulatory networks
>> https://www.biorxiv.org/content/10.1101/2023.05.18.540731v1
Non-Stiff Dynamic Invertible Model of CO-Regulatory Networks (NS-DIMCORN) defines the genetic nexus underpinning specific cellular functions using invertible warping of flexible multivariate Gaussian distributions by neural Ordinary differential equations.
NS-DIMCORN allows unrestricted neural network architectures. NS-DIMCORN represents different cell states by a continuous latent trajectory and defines a bijective map from the latent learned latent space to data by integrating latent variables.
NS-DIMCORN yields a continuous-time invertible generative model with unbiased density estimation by one-pass sampling. NS-DIMCORN achieves easy sampling of the continuous trajectories using Hamiltonian Monte Carlo and calculates nonlinear gene dependency.

□ Protpardelle: An all-atom protein generative model
>> https://www.biorxiv.org/content/10.1101/2023.05.24.542194v1
Protpardelle, an all-atom diffusion model of protein structure, which instantiates a “superposition” over the possible sidechain states, and collapses it to conduct reverse diffusion for sample generation.
Protpardelle is capable of co-designing sequence and structure, it remains a structure-primary generative model that produces estimates of the sequence during its sampling trajectory.
Protpardelle does not define any noising process on the sequence; nor is it a joint model in the sense that we are able to marginalize and condition in some way to produce solutions to the sub-tasks of structure and sequence generation and forward and inverse folding.

□ scGPCL: Deep single-cell RNA-seq data clustering with graph prototypical contrastive learning
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad342/7180270
scGPCL encodes the cell representations based on Graph Neural Networks (GNNs), and utilizes prototypical contrastive learning scheme to learn cell representations by pushing apart semantically disimillar pairs and pulling together similar ones.
scGPCL adopts instance-wise contrastive learning scheme to fully leverage the relational information as well as prototypical contrastive loss to alleviate the limitation of instance-wise contrastive loss.
scGPCL with a cell-gene graph as the input consistently outperforms that w/ a cell-cell graph, which demonstrates that the cell-gene graph better helps to infuse the inherent relational information b/n cells. scGPCL consistently succeeds in learning the cell representation space.

□ scMTNI: Inference of cell type-specific gene regulatory networks on cell lineages from single cell omic datasets
>> https://www.nature.com/articles/s41467-023-38637-9
scMTNI models a GRN as a Dependency network, a probabilistic graphical model with random variables representing genes and regulators, such as transcription factors (TFs) and signaling proteins.
scMTNI’s multi-task learning framework incorporates a probabilistic lineage tree prior. It models the change of a GRN from a start state (e.g., progenitor cell state) to an end state (e.g., more differentiated state) as a series of individual edge-level probabilistic transitions.

□ scSHARP: Consensus Label Propagation with Graph Convolutional Networks for Single-Cell RNA Sequencing Cell Type Annotation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad360/7189733
scSHARP uses a Graph Convolutional Network (GCN) as a mechanism to propagate labels from confidently labeled cells to unconfidently labeled cells. Each GCN used EdgeConv feature propagation between each node and its k closest neighbors, with distance determined dynamically.
scSHARP employes DeepLIFT as an effective Gradient-based interpretation tool for the GCN model. The k hyperparameter and convergence method for the non-parametric neighbor majority approach was chosen with the same validation set used for GCN hyperparameter optimization.

□ IndepthPathway: an integrated tool for in-depth pathway enrichment analysis based on single cell sequencing data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad325/7181277
A Weighted Concept Signature Enrichment Analysis (WCSEA) specialized for pathway enrichment analysis from single cell transcriptomics (scRNA-seq).
WCSEA took a broader approach for assessing the functional relations of pathway gene sets to differentially expressed genes, and leverage the cumulative signature of molecular concepts characteristic of the highly differentially expressed genes.
IndepthPathway presents outstanding stability and depth in pathway enrichment results under stochasticity of the data, thus will substantially improve the scientific rigor of the pathway analysis for single cell sequencing data.

□ ReX: an integrative tool for quantifying and optimizing measurement reliability for the study of individual differences
>> https://www.nature.com/articles/s41592-023-01901-3
Reliability eXplorer (ReX), to facilitate the examination of individual variation and reliability as well as the effective direction for optimization of measuring individual differences in biomarker discovery.
Gradient flows, a two-dimensional field map-based approach to identifying and representing the most effective direction for optimization when measuring individual differences, which is implemented in ReX.

□ Reassessing the modularity of gene co-expression networks using the Stochastic Block Model
>> https://www.biorxiv.org/content/10.1101/2023.05.31.542906v1
The Weighted degree corrected stochastic block model with no free parameters, can find many more gene clusters than competing methods. Second, that such gene clusters are biologically meaningful as revealed by highly specific gene ontology enrichment.
The mean and the variance of the observed edge weights b/n 2 blocks are a function only of the block structure, i.e., genes in the same block have a similar probability of being connected to other genes and the value of the weights in these edges comes from the same distribution.

□ DeepRaccess: High-speed RNA accessibility prediction using deep learning
>> https://www.biorxiv.org/content/10.1101/2023.05.25.542237v1
DeepRaccess, a fast accessibility prediction tool based on deep learning-based software acceleration. DeepRaccess can moderately reproduce the results of Raccess, an existing RNA accessibility calculation method, with high accuracy on both simulation and empirical datasets.
DeepRaccess divides the sequence into subsequences. DeepRaccess predicts the accessibility of these subsequences and integrated them with the accessibility of the full-length RNA. DeepRaccess ignored the accessibility of the 55-base region from the end of each subsequence.

□ Optipyzer: A fast and flexible multi-species codon optimization server
>> https://www.biorxiv.org/content/10.1101/2023.05.22.541759v1
Optipyzer is a new fast and effective multi-species codon optimization server capable of optimizing recombinant DNA sequences for multiple target organisms simultaneously.
Optipyzer leverages the most up-to-date codon usage data through the HIVE-Codon Usage Tables database. The averaged table is used to construct an optimized query using a stochastic selection process and the relative codon adaptation index to ensure a proper expression profile.

□ PatternCode: Design of optimal labeling patterns for optical genome mapping via information theory
>> https://www.biorxiv.org/content/10.1101/2023.05.23.541882v1
An information-theoretic model of optical genome mapping (OGM), which enables the prediction of its accuracy and the design of optimal labeling patterns for specific applications and target organism genomes.
It depends on only four parameters: the target genome length, the DNA fragment length, and two easily estimated parameters: the label detection likelihood, estimated from experimental genome-aligned DNA fragment images, and the labeling pattern distribution.
This enables the design of better OGM experiments, and allows for the intuitive understanding of the importance of different parameters on the accuracy, such as the logarithmic dependence on the target genome length versus the polynomial dependence on the fragment length.
Additionally, the model enables fast computation due to its simple analytical form, allowing for the design of protocols where multiple patterns are labeled with multiple labeling reagents through combinatorial optimization of pattern combination selection.

□ On the invariant subspace problem in Hilbert spaces
>> https://arxiv.org/abs/2305.15442
Every bounded linear operator T on a Hilbert space H has a closed non-trivial invariant subspace. There are situations when we cannot just use the Main Construction to reach a non-cyclic vector.
If we had norm convergence we could continue beyond (εθ)'. We "get stuck" if (am), (bm) (Ko-1, (kim)m»1) become close to being linearly dependent. We create εθ's arbitrarily near to 0, from which we restart the Main Construction.

□ MEGABYTE: Predicting Million-byte Sequences with Multiscale Transformers
>> https://arxiv.org/abs/2305.07185
MEGABYTE is an autoregressive model for efficiently modeling long input sequences. MEGABYTE is able to handle all sequence lengths with a single forward pass of up to 1.2M tokens.
MEGABYTE uses an efficient decoder model by using a intra-patch transformer to predict each sequence element's likelihood, and offseting the inputs to the two models to avoid leaking information.

□ cPeaks: Consensus peaks of chromatin accessibility in the human genome
>> https://www.biorxiv.org/content/10.1101/2023.05.30.542889v1
Predicting all potential open regions in the human genome using cPeaks. It can be regarded as a new set of epigenomic elements in the human genome. cPeaks also have the potential to identify rare cell subtypes that are difficult to be detected using pseudo-bulk peaks.
Each approach provided a genomic region set as a reference for mapping sequencing reads to generate a cell-by-chromatin accessibility feature matrix. cPeaks got similar or better performance in comparison with other feature-defining approaches under all evaluation methods.

□ RaggedExperiment: the missing link between genomic ranges and matrices in Bioconductor
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad330/7174143
RaggedExperiment represents ragged genomic ranges from multiple samples, and to provide flexible and efficient tools for matrix-format summarization across identical ranges in each sample.
RaggedExperiment fills a gap in providing efficient, flexible conversion between "ragged" genomic data and matrix format for which we are not aware of a direct analogy to benchmark against.

□ BRGenomics for analyzing high resolution genomics data in R
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad331/7174141
BRGenomics provides various methods for data importation and processing, read counting and aggregation, spike-in and batch normalization, re-sampling methods for robust “metagene” analyses, and various other functions for cleaning and modifying sequencing and annotation data.
BRGenomics has been used to analyze ATAC-seq, ChIP- seq/ChIP-exo, PRO-seq/PRO-cap, and RNA-seq data; is built to be unobtrusive and maximally compatible with the Bioconductor ecosystem.

□ Matías Gutiérrez
Here’s what coming #NanoporeConf @nanopore

□ GraphSNP: an interactive distance viewer for investigating outbreaks and transmission networks using a graph approach
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05332-x
GraphSNP is an interactive visualisation tool running in a web browser that allows users to rapidly generate pairwise SNP distance networks, investigate SNP distance distributions, identify clusters of related organisms, and reconstruct transmission routes.
GraphSNP generates pairwise Hamming distance from the SNP alignment. GraphSNP provides capability for creating a Minimum Spanning Tree of the resulted clusters using the Kruskal’s algorithm, a transmission tree using the SeqTrack algorithm, and the breadth-first search algorithm.

□ Adversarial training improves model interpretability in single-cell RNA-seq analysis
>> https://www.biorxiv.org/content/10.1101/2023.05.17.541170v1
Adversarial training fortifies a deep learning model, which can be useful for future clinical and health applications, such as diagnostic or prognostic gene expression biomarkers or patient classification, that need to be robust against adversarial attacks.
Projected Gradient Descent (PGD) and Fast Gradient Signed Method (FGSM). These take the trained model and introduce noise in the input data in the direction of the model gradient that has the greatest impact on the model's accuracy.

□ iDeLUCS: A deep learning interactive tool for alignment-free clustering of DNA sequences
>> https://www.biorxiv.org/content/10.1101/2023.05.17.541163v1
iDeLUCS is a standalone software tool that exploits the capabilities of deep learning to cluster genomic sequences. It is agnostic to the data source, making it suitable for genomic sequences taken from any organism in any kingdom of life.
iDeLUCS assigns a cluster identifier to every DNA sequence present in a dataset, while incorporating several built-in visualization tools that provide insights into the underlying training process. iDeLUCS offers an evaluation mode to compare the ground-truth label assignments.
This is accompanied by a visual qualitative assessment of the clustering, through the use of the uniform manifold approximation of the learned lower dimensional embedding. iDeLUCS outputs confidence scores for all of its cluster-label predictions, for enhanced interpretability.

□ Genome Context Viewer (GCV) version 2: enhanced visual exploration of multiple annotated genomes
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkad391/7173788
Version 2 of the Genome Context Viewer (GCV) – an open-source web application that uses the functional annotations of genes to perform on-demand federated synteny analysis of collections of genomes.
By using functional annotations as the unit of search and comparison, GCV can compute and display multiple regions across several assemblies from different databases in real-time.

□ Benchtop DNA printers are coming soon—and biosecurity experts are worried
>>
https://www.science.org/content/article/benchtop-dna-printers-are-coming-soon-and-biosecurity-experts-are-worried
The current screening system, which is voluntary, “could be upended by benchtop DNA synthesis,” says report co-author Jaime Yassif, vice president for global biological policy and programs at the Nuclear Threat Initiative.
The report recommends that benchtop synthesis devicemakers vet their customers to ensure they are legitimate biotechnology researchers. It also calls for build-in protections, such as software that allows the manufacturer to screen all requests for DNA sequences prior to synthesis.

□ SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms
>> https://www.biorxiv.org/content/10.1101/2023.05.17.541248v1
SQANTI3 provides an extensive naming framework to characterize transcript model diversity. The incorporates novel metrics and features to better characterize the transcription start and end sites, splice junctions of isoforms, and filter out potential artifacts.
The Rescue module re-evaluates artifacts to suggest a bona fide replacement transcript model and avoid the loss of known genes and transcripts for which evidence of expression exists.
SQANTI3 includes a Random Forest classifier that labels long read transcripts as isoforms or artifacts using SQANTI QC descriptors as predictive variables and a set of user-defined true and false transcripts.

□ Unsupervised single-cell clustering with Asymmetric Within-Sample Transformation and per cluster supervised features selection
>> https://www.biorxiv.org/content/10.1101/2023.05.17.541148v1
The asymmetric transformation is a special winsorization that flattens low-expressed intensities and preserves highly expressed gene levels. An intermediate step removes non-informative genes according to a threshold applied to a per-gene entropy estimate.
Following the clustering, a time-intensive algorithm is shown to uncover the molecular features associated with each cluster. This step implements a resampling algorithm to generate a random baseline to measure up/down-regulated significant genes.
□ 『遺伝情報は誰のものか』問題。DNAは個人の資産か、公衆衛生やセキュリティリスクを内包する資源とするか
□ Mike White QT
>> https://twitter.com/genologos/status/1660414328439287810?s=61&t=YtYFeKCMJNEmL5uKc0oPFg
I’ve never been able to follow this reasoning. If an individual with substantial Native American ancestry wants to contribute their DNA to a genomics project, do they need to get permission from some tribal authority that this person may not even acknowledge?

□ SeATAC: a tool for exploring the chromatin landscape and the role of pioneer factors
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02954-5
SeATAC uses a conditional variational autoencoder model to learn the latent representation of ATAC-seq V-plots and outperforms MACS2 and NucleoATAC on six separate tasks.
The SeATAC model uses a V-plot with a width of 640-bp genomic region and a height of 640 bp of fragment sizes that covers nucleosome free reads, mono-nucleosome reads, di-nucleosome reads, and tri-nucleosomes.

□ In silico methods for predicting functional synonymous variants
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02966-1
Genscan uses a maximal dependence decomposition (MDD) model, which is a decision tree-based method. Genesplicer combines MDD with Markov models (MM) to capture additional dependencies between neighboring positions.
MES uses maximum entropy principle (MEP) for modeling short sequence motifs found in splice sites while also accounting for higher-order dependencies between adjacent and non-adjacent positions.
usDSM (Deleterious Synonymous Mutation Prediction using Undersampling Scheme) and synVep (Synonymous Variant Effect Predictor) are newer tools that have demonstrated improved proficiencies by implementing undersampling methods and positive-unlabeled learning.

□ GoldRush: Linear time complexity de novo long read genome assembly
>> https://www.nature.com/articles/s41467-023-38716-x
GoldRush, a memory-efficient long-read haploid de novo genome assembler that employs a novel long-read assembly algorithm, which runs in linear time in the number of reads.
GoldPath iterates through the reads, querying each read against a dynamic and probabilistic multi-index Bloom filter data structure in turn, and inserts selected sequence or skips over the read depending on the results of the query to generate multiple silver paths.
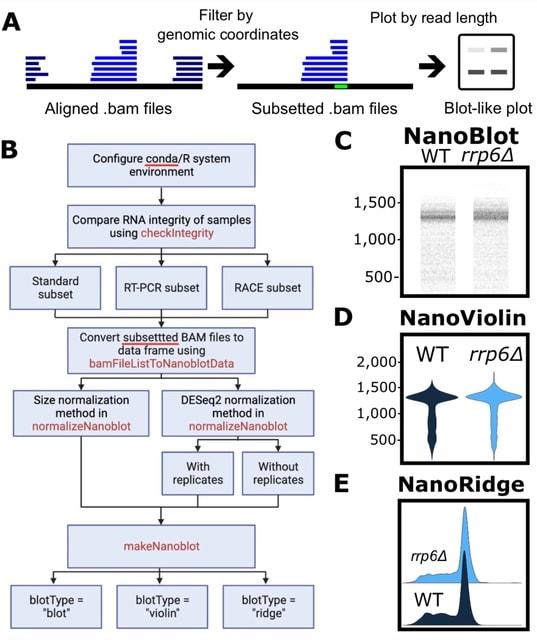
□ NanoBlot: An R-Package for Visualization of RNA Isoforms from Long Read RNA-sequencing Data
>> https://rnajournal.cshlp.org/content/early/2023/05/03/rna.079505.122.abstract
NanoBlot, an open-source, R-package, which generates northern blot and RT-PCR-like images from long-read sequencing data. NanoBlot requires aligned, positionally sorted and indexed BAM files.
NanoBlot can output other visualizations such as violin plots and 3′-RACE-like plots focused on 3′-ends isoforms visualization. The use of the NanoBlot package should provide a simple answer to some of the challenges of visualizing long-read RNA sequencing data.










※コメント投稿者のブログIDはブログ作成者のみに通知されます