□ Scalable network reconstruction in subquadratic time
>> https://arxiv.org/abs/2401.01404
A general algorithm applicable to a broad range of reconstruction problems that achieves its result in subquadratic time, with a data-dependent complexity loosely upper bounded by O(N3/2 log N), but with a more typical log-linear complexity of O(N log2 N).
This algorithm relies on a stochastic second neighbor search that produces the best edge candidates with high probability, thus bypassing an exhaustive quadratic search.
This algorithm achieves a performance that is many orders of magnitude faster than the quadratic baseline, allows for easy parallelization. The strategy is applicable for algorithms that can be used w/ non-convex objectives, e.g. stochastic gradient descent / simulated annealing.

□ OmniNA: A foundation model for nucleotide sequences
>> https://www.biorxiv.org/content/10.1101/2024.01.14.575543v1
OmniNA represents an endeavor in leveraging foundation models for comprehensive nucleotide learning across diverse species and genome contexts. OmniNA can be fine-tuned to align multiple nucleotide learning tasks with natural language paradigms.
OmniNA employs a transformer-based decoder, undergoes pre-training through an auto-regressive approach. OmniNA was pre-trained on a scale of 91.7 million nucleotide sequences encompassing 1076.2 billion bases range across a global species and biological context.

□ STIGMA: Single-cell tissue-specific gene prioritization using machine learning
>> https://www.sciencedirect.com/science/article/pii/S0002929723004433
STIGMA predicts the disease-causing probability of genes based on their expression profiles across cell types, while considering the temporal dynamics during the embryogenesis of a healthy (wild-type) organism, as well as several intrinsic gene properties.
In STIGMA, supervised machine learning is applied to the single-cell gene expression data as well as intrinsic gene properties on positive and negative classes.
The STIGMA score that each gene receives is based on the cell type-specific temporal dynamics in gene expression and, to a smaller extent, is based on the gene-intrinsic metrics, including the population level constraint metrics.

□ RfamGen: Deep generative design of RNA family sequences
>> https://www.nature.com/articles/s41592-023-02148-8
RfamGen (RNA family sequence generator), a deep generative model that designs RNA family sequences in a data-efficient manner by explicitly incorporating alignment and consensus secondary structure information.
RfamGen can generate novel and functional RNA family sequences by sampling points from a semantically rich and continuous representation. RfamGen successfully generates artificial sequences with higher activity than natural sequences.

□ SYNTERUPTOR: mining genomic islands for non-classical specialised metabolite gene clusters
>> https://www.biorxiv.org/content/10.1101/2024.01.03.573040v1
SYNTERUPTR identifies genomic islands in a given genome by comparing its genomic sequence with those of closely related species. SYNTERUPTOR was designed and is focused on identifying SMBGC-containing genomic islands.
SYNTERUPTOR pipeline requires a dataset consisting of genome files selected by the user from species that are related enough to possess synteny blocks.
SYNTERUPTOR proceeds by performing pairwise comparisons between all Coding DNA Sequences (CDSs) amino acid sequences to identify orthologs. Subsequently, it constructs synteny blocks and detects any instances of synteny breaks.

□ ALG-DDI: A multi-scale feature fusion model based on biological knowledge graph and transformer-encoder for drug-drug interaction prediction
>> https://www.biorxiv.org/content/10.1101/2024.01.12.575305v1
ALG-DDI can comprehensively incorporate attribute information, local biological information, and global semantic information. ALG-DDI first employs the Attribute Masking method to obtain the embedding vector of the molecular graph.
ALG-DDI leverages heterogeneous graphs to capture the local biological information between drugs and several highly related biological entities. The global semantic information is also learned from the medicine-oriented large knowledge graphs.
ALG-DDI employs a transformer encoder to fuse the multi-scale drug representations and feed the resulting drug pair vector into a fully connected neural network for prediction.

□ FAVA: High-quality functional association networks inferred from scRNA-seq and proteomics data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae010/7513163
FAVA (Functional Associations using Variational Autoencoders) compresses high-dimensional data into a low-dimensional space. FAVA infers networks from high-dimensional omics data with much higher accuracy, across a diverse collection of real as well as simulated datasets.
In latent space, FAVA calculates the Pearson correlation coefficient (PCC) each pair of proteins, resulting in a functional association network. FAVA can process large datasets w/ over 0.5 million conditions and has predicted 4,210 interactions b/n 1,039 understudied proteins.

□ FFS: Fractal feature selection model for enhancing high-dimensional biological problems
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05619-z
In fractals, a central tenet posits that patterns recur at differing scales. This principle suggests that when one examines a minuscule segment of a fractal and juxtaposes it with a more significant portion of the same fractal, the patterns observed will bear striking resemblance.
FFS (Fractal Feature Selection) is proof of harmonic convergence of a low-complexity system with remarkable performance. FFS partitions features into blocks, measures similarity using the Root Mean Square Error (RMSE), and determines feature importance based on low RMSE values.
By conceptualizing these attributes as blocks, where each block corresponds to a particular data category, the proposed model finds that blocks with common similarities are often associated with specific data categories.

□ CytoCommunity: Unsupervised and supervised discovery of tissue cellular neighborhoods from cell phenotypes
>> https://www.nature.com/articles/s41592-023-02124-2
CytoCommunity learns a mapping directly from the cell phenotype space to the TCN space using a graph neural network model without intermediate clustering of cell embeddings.
By leveraging graph pooling, CytoCommunity enables de novo identification of condition-specific and predictive TCNs under the supervision of sample labels.
CytoCommunity formulates TCN identification as a community detection problem on graphs and use a graph minimum cut (MinCut)-based GNN model to identify TCNs.
CytoCommunity directly uses cell phenotypes as features to learn TCN partitions and thus facilitates the interpretation of TCN functions.
CytoCommunity can also identify condition-specific TCNs from a cohort of labeled tissue samples by leveraging differentiable graph pooling and sample labels, which is an effective strategy to address the difficulty of graph alignment.

□ scSNV-seq: high-throughput phenotyping of single nucleotide variants by coupled single-cell genotyping and transcriptomics
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03169-y
scSNV-seq uses transcribed genetic barcodes to couple targeted single-cell genotyping with transcriptomics to identify the edited genotype and transcriptome of each individual cell rather than predicting genotype from gRNA identity.
scSNV-seq allows us to identify benign variants or variants with an intermediate phenotype which would otherwise not be possible.
The methodology is applicable to any other methods for introducing variation such as HDR, prime editing, or saturation genome editing since it does not rely on gRNA identity to infer genotype.

□ Fragmentstein: Facilitating data reuse for cell-free DNA fragment analysis
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae017/7550024
Fragmentstein, a command-line tool for converting non-sensitive cDNA-fragmentation data into alignment mapping (BAM) files. Fragmentstein complements fragment coordinates with sequence information from a reference genome to reconstruct BAM files.
Fragmentstein creates alignment files for each sample using only non-sensitive information. The original alignment files and the alignment files generated by Fragmentstein were subjected to fragment length, copy number and nucleosome occupancy analysis.
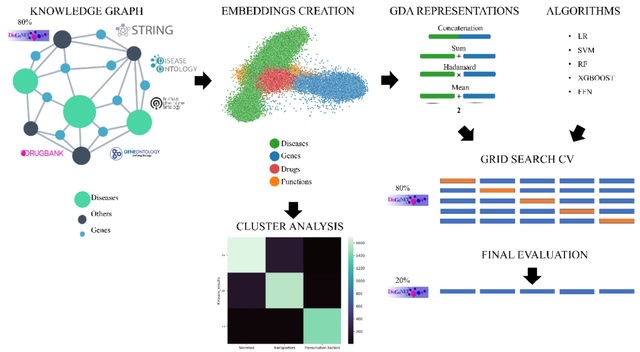
□ DLemb / BioKG2Vec: PREDICTING GENE DISEASE ASSOCIATIONS WITH KNOWLEDGE GRAPH EMBEDDINGS FOR DISEASES WITH CURTAILED INFORMATION
>> https://www.biorxiv.org/content/10.1101/2024.01.11.575314v1
BioKg2Vec relies on a biased random-walk approach in which the user can prioritize specific connections by assigning a weight to edges. In the KG defined in this work we used 4 different node-types: drug, protein, function and disease.
DLemb is a shallow neural network. The input layer takes as input KG entities as numbers and outputs them to the embedding layer. Subsequently, embeddings are normalized, and a dot product is calculated between them resulting in the output layer.
DLemb is trained by providing a batch of correct links and wrong links in the KG to provide with positive and negative examples in what can be conceived as a link-prediction task. Embeddings are then optimized for every epoch by minimizing RMSE and using Adam optimization.

□ POP-GWAS: Valid inference for machine learning-assisted GWAS
>> https://www.medrxiv.org/content/10.1101/2024.01.03.24300779v1
POP-GWAS (Post-prediction GWAS) provides unbiased estimates and well-calibrated type-l error, is universally more powerful than conventional GWAS on the observed phenotype, and has minimal assumption on the variables used for imputation and choice of prediction algorithm.
POP-GWAS imputes the phenotype in both labeled and unlabeled samples, and performs three GWAS: GWAS of the observed and imputed phenotype in labeled samples, and GWAS on the imputed phenotype in unlabeled samples.

□ GLDADec: marker-gene guided LDA modelling for bulk gene expression deconvolution
>> https://www.biorxiv.org/content/10.1101/2024.01.08.574749v1
GLADADec (Guided Latent Dirichlet Allocation Deconvolution) utilizes marker gene names as partial prior information to estimate cell type proportions, thereby overcoming the challenges of conventional reference-based and reference-free methods simultaneously.
GLADADec employs a semi-supervised learning algorithm that combines cell-type marker genes with additional factors that may influence gene expression profiles to achieve a robust estimation of cell type proportions. An ensemble strategy is used to aggregate the output.

□ scGOclust: leveraging gene ontology to compare cell types across distant species using scRNA-seq data
>> https://www.biorxiv.org/content/10.1101/2024.01.09.574675v1
scGOclust constructs a functional profile of individual cells by multiplication of a gene expression count matrix of cells and a binary matrix with GO BP annotations of genes.
This GO BP feature matrix is treated similarly to a count matrix in classic single-cell RNA sequencing (scRNA-seq) analysis and is subjected to dimensionality reduction and clustering analyses.
scGOclust recapitulates the function spectrum of different cell types, characterises functional similarities between homologous cell types, and reveals functional convergence between unrelated cell types.

□ MATES: A Deep Learning-Based Model for Locus-specific Quantification of Transposable Elements in Single Cell
>> https://www.biorxiv.org/content/10.1101/2024.01.09.574909v1
MATES (Multi-mapping Alignment for TE loci quantification in Single-cell), a novel deep neural network-based method tailored for accurate locus-specific TE quantification in single-cell sequencing data across modalities.
MATES harnesses the distribution of uniquely mapped reads occurrence flanking TE loci and assigns multiple mapping TE reads for locus-specific TE quantification.
MATES captures complex relationships b/n the context distribution of unique-mapping reads flanking TE loci and the probability of multi-mapping reads assigned to those loci, handles the multi-mapping read assignments probabilistically based on the local context of the TE loci.

□ COFFEE: CONSENSUS SINGLE CELL-TYPE SPECIFIC INFERENCE FOR GENE REGULATORY NETWORKS
>> https://www.biorxiv.org/content/10.1101/2024.01.05.574445v1
COFFEE (COnsensus single cell-type speciFic inFerence for gEnE regulatory networks), a Borda voting based consensus algorithm that integrates information from 10 established GRN inference methods.
COFFEE has improved performance across synthetic, curated and experimental datasets when compared to baseline methods.
COFFEE's stability across differing datasets; even with Curated data, the consensus approach is able to capture high confidence edges when compared to the ground truth data.

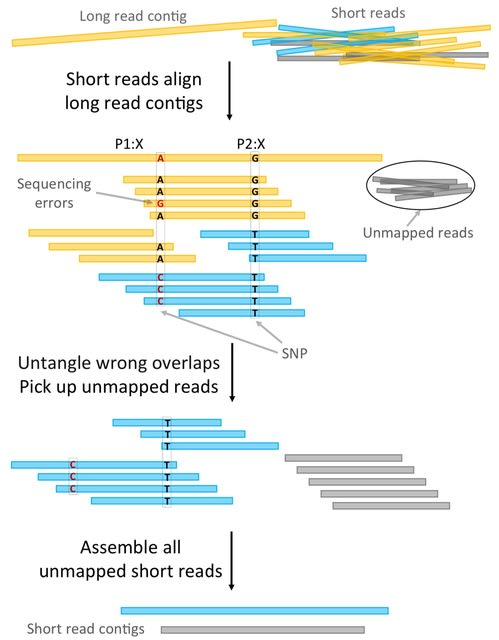
□ HAT: de novo variant calling for highly accurate short-read and long-read sequencing data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad775/7510834
Hare And Tortoise (HAT) as an automated DNV detection workflow for highly accurate short-read and long-read sequencing data.
HAT is a computational workflow that begins with aligned read data (i.e., CRAM or BAM) from a parent-child sequenced trio and outputs DNVs. The HAT workflow consists of three main steps: GVCF generation, fam-ily-level genotyping, and filtering of variants to get final DNVs.
HAT detects high-quality DNVs from Illumina short-read whole-exome sequencing, Illumina short-read whole-genome sequencing, and highly accurate PacBio HiFi long-read whole-genome sequencing data.
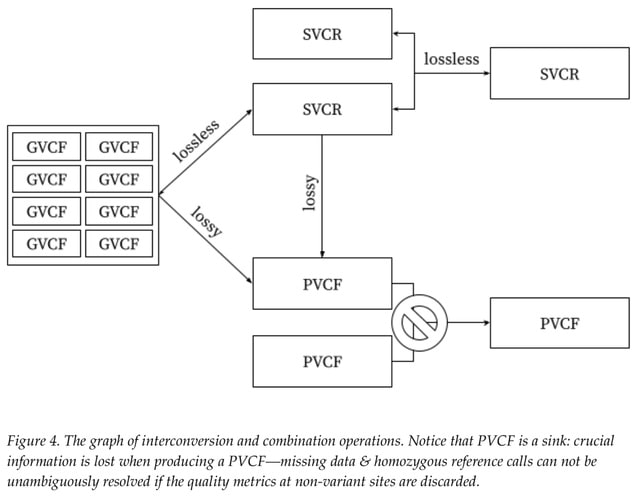
□ SVCR: The Scalable Variant Call Representation: Enabling Genetic Analysis Beyond One Million Genomes
>> https://www.biorxiv.org/content/10.1101/2024.01.09.574205v1
SVCR achieves this by adopting reference blocks from the Genomic Variant Call Format (GVCF) and employing local allele indices. SVCR is also lossless and mergeable, allowing for N+1 and N+K incremental joint-calling.
SVCR-VCF encodes SVCR in VCF format, and VDS, which uses Hail's native format. Their experiments confirm the linear scalability of SVCR-VCF and VDS, in contrast to the super-linear growth seen with standard VCF files.
VDS Combiner, a scalable, open-source tool for producing a VDS from GVCFs and unique features of VDS which enable rapid data analysis.
PVCF defines the semantics of fields such as GT, AD, GP, PL, and, for list fields, the relationship between their length and the number of alternate alleles. VCF, as a format, describes, for example, how a number or a list is rendered in plaintext.
PVCF represents a collection of sequences as a dense matrix, with one column per sequenced sample and one row for every variant site. PVCF permits both a multiallelic representation (wherein each locus appears in at most one row) and a biallelic representation.
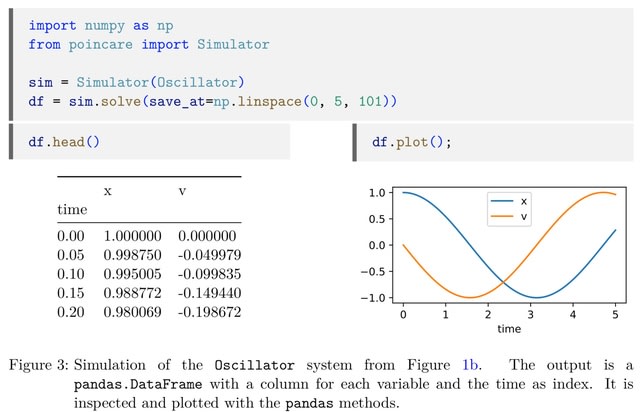
□ Poincaré and SimBio: a versatile and extensible Python ecosystem for modeling systems.
>> https://www.biorxiv.org/content/10.1101/2024.01.10.574883v1
Poincaré allows defining differential equation sys-tems, while SimBio builds on it for defining reaction networks. They are focused on providing an ergonomic experience to end-users by integrating well with IDEs and static analysis tools through the use of standard modern Python syntax.
The models built using these packages can be introspected to create other representations, such as graphs connecting species and/or reactions, or tables with parameters or equations.

□ Secreted Particle Information Transfer (SPIT) - A Cellular Platform For In Vivo Genetic Engineering
>> https://www.biorxiv.org/content/10.1101/2024.01.11.575257v1
Compared to the limited packaging capacities of contemporary in vivo gene therapy delivery platforms, a human cell's nucleus contains approximately 6 billion base pairs of information. They hypothesized that human cells could be applied as vectors for in vivo gene therapy.
SPIT is modified to secrete a genetic engineering enzyme within a particle that transfers this enzyme into a recipient cell, where it manipulates genetic information.

□ Decoder-seq enhances mRNA capture efficiency in spatial RNA sequencing
>> https://www.nature.com/articles/s41587-023-02086-y
Decoder-seq (Dendrimeric DNA coordinate barcoding design for spatial RNA sequencing) combines dendrimeric nanosubstrates with microfluidic coordinate barcoding to generate spatial arrays with a DNA density approximately ten times higher than previously reported methods.
Decoder-seq improves the detection of lowly expressed olfactory receptor (Olfr) genes in mouse olfactory bulbs and contributed to the discovery of a unique layer enrichment pattern for two Olfr genes.

□ GVRP: Genome Variant Refinement Pipeline for variant analysis in non-human species using machine learning
>> https://www.biorxiv.org/content/10.1101/2024.01.14.575595v1
GVRP employs a machine learning-based approach to refine variant calls in non-human species. Rather than training separate variant callers for each species, we employ a machine learning model to accurately identify variations and filter out false positives from DeepVariant.
In GVRP, they omit certain DeepVariant preprocessing steps and leverage the ground-truth Genome In A Bottle (GIAB) variant calls to train the machine learning model for non-human species genome variant refinement.

□ BAMBI: Integrative biostatistical and artificial-intelligence method discover coding and non-coding RNA genes as biomarkers
>> https://www.biorxiv.org/content/10.1101/2024.01.12.575460v1
BAMBI (Biostatistics and Artificial-Intelligence integrated Method for Biomarker /dentification), a robust pipeline that identifies both coding and non-coding RNA biomarkers for disease diagnosis and prognosis.
BAMBI can process RNA-seq data and microarray data to pinpoint a minimal yet highly predictive set of RNA biomarkers, thus facilitating their clinical application.
BAMBI offers visualization of biomarker expression and interpretation their functions using co-expression networks and literature mining, enhancing the interpretability of the results.

□ PoMoCNV: Inferring the selective history of CNVs using a maximum likelihood model
>> https://www.biorxiv.org/content/10.1101/2024.01.15.575676v1
PoMoCNV (POlymorphism-aware phylogenetic MOdel for CNV datasets) infers the fitness parameters and transition rates associated with different copy numbers along branches in the phylogenetic tree, tracing back in time.
Utilizing the phylogenetic tree of populations and estimated copy numbers, PoMoCNV was utilized to infer the evolutionary parameters governing CNV evolution along branches.
In PoMoCNV, the likelihood of this birth-death process is modeled per genomic segment, taking into account the copy number (allele) fitness and frequencies.

□ O-LGT: Online Hybrid Neural Network for Stock Price Prediction: A Case Study of High-Frequency Stock Trading in the Chinese Market
>> https://www.mdpi.com/2225-1146/11/2/13
O-LGT, an online hybrid recurrent neural network model tailored for analyzing LOB data and predicting stock price fluctuations in a high-frequency trading (HFT) environment.
O-LGT combines LSTM, GRU, and transformer layers, and features efficient storage management. When computing the stock forecast for the immediate future, O-LGT only use the output calculated from the previous trading data together with the current trading data.

□ GYOSA: A Distributed Computing Solution for Privacy-Preserving Genome-Wide Association Studies
>> https://www.biorxiv.org/content/10.1101/2024.01.15.575678v1
GYOSA, a secure and privacy-preserving distributed genomic analysis solution. Unlike in previous work, GYOSA follows a distributed processing design that enables handling larger amounts of genomic data in a scalable and efficient fashion.
GYOSA provides transparent authenticated encryption, which protects sensitive data from being disclosed to unwanted parties and ensures anti-tampering properties for clients' data stored in untrusted infrastructures.

□ KaMRaT: a C++ toolkit for k-mer count matrix dimension reduction
>> https://www.biorxiv.org/content/10.1101/2024.01.15.575511v1
KaMRaT (k-mer Matrix Reduction Toolkit) is a program for processing large k-mer count tables extracted from high throughput sequencing data.
Major functions include scoring k-mers based on count statistics, merging overlapping k-mers into longer contigs and selecting k-mers based on their presence in certain samples.
KaMRaT merge builds on the concept of local k-mer extension ("unitigs") to improve extension precision by leveraging count data. KaMRaT enables the identification of condition-specific or differential sequences, irrespective of any gene or transcript annotation.

□ EvoAug-TF: Extending evolution-inspired data augmentations for genomic deep learning to TensorFlow
>> https://www.biorxiv.org/content/10.1101/2024.01.17.575961v1
EvoAug was introduced to train a genomic DNN with evolution-inspired augmentations. EvoAug-trained DNNs have demonstrated improved generalization and interpretability with attribution analysis.
EvoAug-TF is a TensorFlow implementation of EvoAug (a PyTorch package) that provides the ability to train genomic DNNs with evolution-inspired data augmentations. EvoAug-TF improves generalization and model interpretability with attribution methods.

□ SLEDGe: Inference of ancient whole genome duplications using machine learning
>> https://www.biorxiv.org/content/10.1101/2024.01.17.574559v1
SLEDGe (Supervised Learning Estimation of Duplicated Genomes) provides a novel means to repeatably and rapidly infer ancient WGD events
from Ks plots derived from genomic or transcriptomic data.
SLEDGe can simulate ancient WGDs of multiple ages and across a range of gene birth and death rates. It provides the first model-based approach to infer WGDs in Ks plots and makes WGD interpretation more repeatable and consistent.
□ Peter Kochinsky
>> https://rapport.bio/all-stories/semper-maior-spirits-rising-january-2024
Do you think of biotech as wasteful? How much of the biotech Universe's cash is locked away in companies that have lingered all year with a negative enterprise value? We looked.
Interested in the relevance of M&A to sector returns? How much of the returns from M&A accrue to companies held by at least one specialist? At least three? We looked.
What's it all mean for private companies looking to get public?
And overshadowing it all is a question: what can we do to protect the @biotech sector and biomedical innovation from the wrong stroke of a pen?

 (Created with Midjourney v6.0 ALPHA)
(Created with Midjourney v6.0 ALPHA)