□ PCA-Plus: Enhanced principal component analysis with illustrative applications to batch effects and their quantitation
>> https://www.biorxiv.org/content/10.1101/2024.01.02.573793v1
DSC (the dispersion separability criterion), a novel variant metric for quantifying the global dissimilarity of sets of pre-defined groups, with application to PCA plots.
The DSC can be used, for instance, to assess the magnitude of batch effects or the differences among classes or subtypes of biological samples.
PCA-Plus features group centroids; trend arrows (when pertinent); separate coloring of centroids, rays, and data points; and quantitation in terms of the new DSC metric with corresponding permutation test p-values.
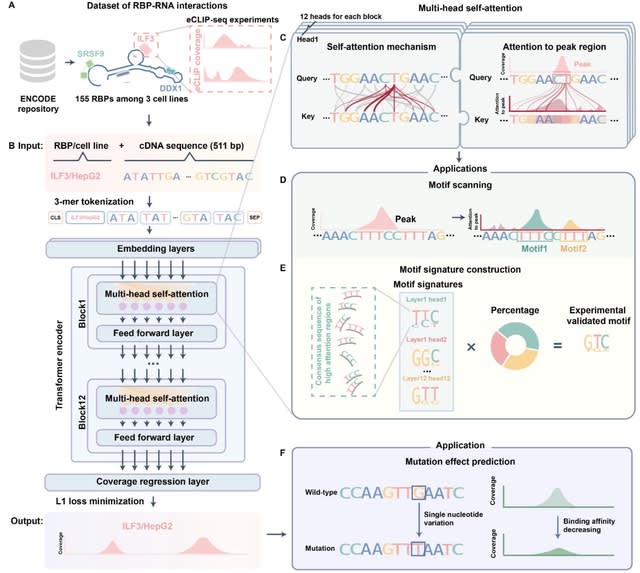
□ Reformer: Deep learning model for characterizing protein-RNA interactions from sequence at single-base resolution
>> https://www.biorxiv.org/content/10.1101/2024.01.14.575540v1
Reformer is based on transformer aiming to improve prediction resolution and facilitate greater information flow between peaks and their surrounding contexts.
Reformer provides a unified framework for characterizing RBP binding and prioritizing mutations that affect RNA regulation at base resolution. For each base, the transformer layer computed a weighted sum across the representations of all other bases of the sequence.
Reformer refines predictions by incorporating information from relevant regions across the entire sequence. Employing a regression layer for coverage prediction, Reformer outputs binding affinities for all bases.
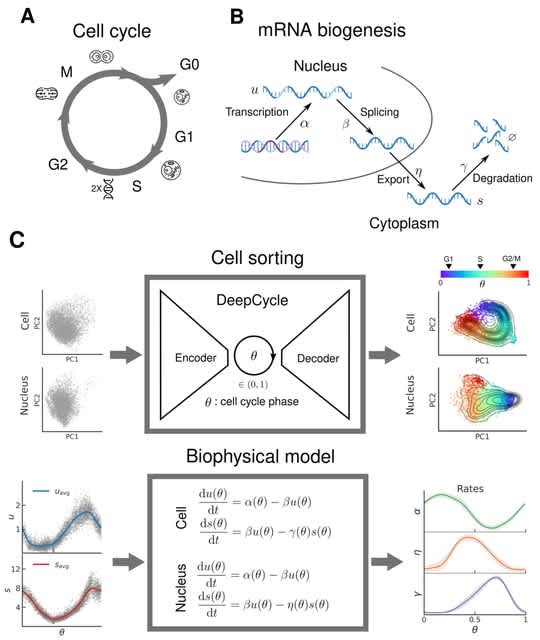
□ DeepCycle: Unraveling the oscillatory dynamics of mRNA metabolism and chromatin accessibility during the cell cycle through integration of single-cell multiomic data
>> https://www.biorxiv.org/content/10.1101/2024.01.11.575159v1
DeepCycle, a deep learning tool that uses single-cell RNA sequencing, to map the gene expression profiles of every cell to a continuous latent variable, 0, representing the cell cycle phase.
DeepCycle predicts the cell cycle dependence of transcription, nuclear export, and degradation rates for every gene, revealing waves of transcriptional and post-transcriptional regulation during the cell cycle.
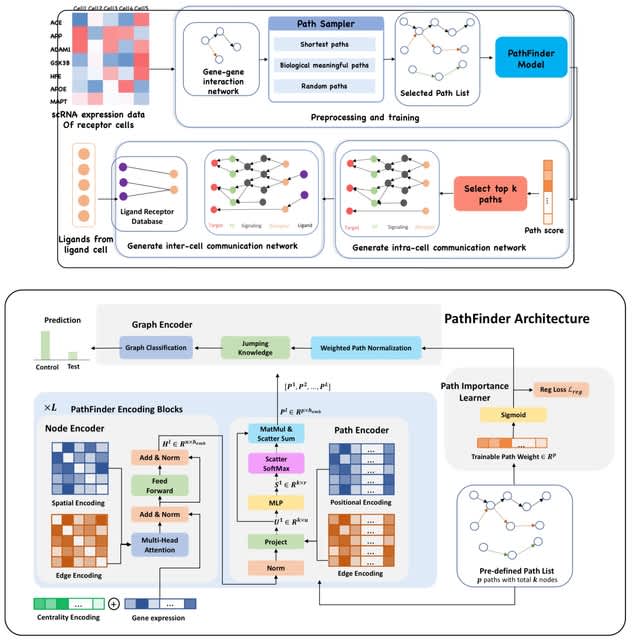
□ PathFinder: a novel graph transformer model to infer multi-cell intra- and inter-cellular signaling pathways and communications
>> https://www.biorxiv.org/content/10.1101/2024.01.13.575534v1
PathFinder is based on the divide-and-conquer strategy, which divides the complex signaling networks into signaling paths, and then score and rank them using a novel graph transformer architecture to infer the intra- and inter-cell signaling network inference.
PathFinder can effectively separate cells from different conditions by selecting differentially expressed signaling paths. The trainable path weight will be learned to assign each path an importance score, which can be used to generate intra-cell communication networks.

□ scKWARN: Kernel-weighted-average robust normalization for single-cell RNA-seq data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae008/7574580
scKWARN, a Kernel Weighted Average Robust Normalization designed to correct known or hidden technical cofounders w/o assuming specific data distributions or count-depth relationships. scKWARN inherently consider any technical factors contributing to unwanted expression variation.
scKWARN generates a pseudo expression profile for EA cell using information from its fuzzy technical neighbors through a kernel smoother. It then compares this profile against the reference derived from cells w/ the same bimodality patterns to determine the normalization factor.
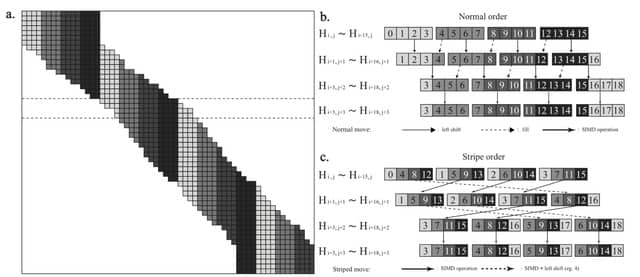
□ BSAlign: a library for nucleotide sequence alignment
>> https://www.biorxiv.org/content/10.1101/2024.01.15.575791v1
BSalign is a library/tool for adaptive banding striped 8/2-bit-scoring global/extend/overlap DNA sequence pairwise/multiple alignment
BSAlign delivers alignment results at an ultra-fast speed by knitting a series of novel methods together to take advantage of all of the aforementioned three perspectives w/ highlights such as active F-loop in striped vectorization and striped move in banded dynamic programming.

□ SI: Quantifying the distribution of feature values over data represented in arbitrary dimensional spaces
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011768
Structure Index (SI), a new metric aimed at quantifying how a given feature is structured along an arbitrary point cloud. The SI aims at quantifying the amount of structure present in the distribution of a given feature over a point cloud in an arbitrary D-dimensional space.
By definition, the SI is agnostic to the type of structure (e.g., gradient, patchy, etc.) since bin groups do not need to follow any specific arrangement. SI permits examination of the local and global distribution of features, whether categorical/continuous or scalar/vectorial.
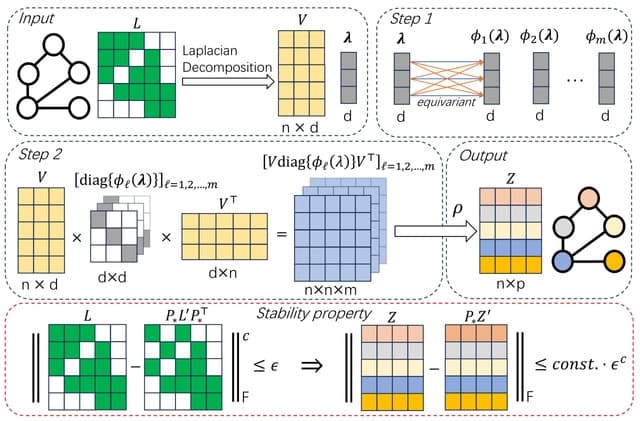
□ SPE: On the Stability of Expressive Positional Encodings for Graph Neural Networks
>> https://arxiv.org/abs/2310.02579
Stable and Expressive Positional Encodings (SPE), an architecture for processing eigenvectors that uses eigenvalues to "softly partition" eigenspaces.
SPE is the first architecture that is provably stable, and universally expressive for basis invariant functions whilst respecting all symmetries of eigenvectors.
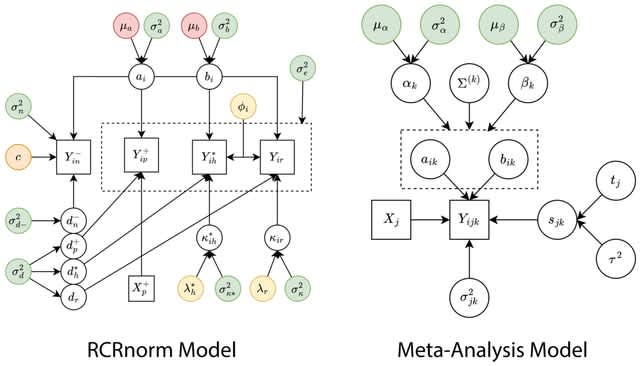
□ MetaNorm: Incorporating meta-analytic priors into normalization of NanoString nCounter data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae024/7574576
MetaNorm, a Bayesian algorithm for normalizing NanoString nCounter gene expression data. performance. MetaNorm employs priors carefully constructed from a rigorous meta- analysis to leverage information.
MetaNorm is based on RCRnorm, a powerful method designed under an integrated series of hierarchical models that allow various sources of error to be explained by different types of probes in the nCounter system.
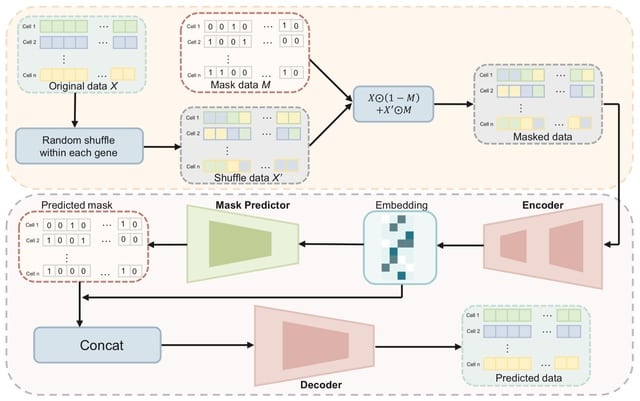
□ scMAE: a masked autoencoder for single-cell RNA-seq clustering
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae020/7564641
scMAE perturbs gene expression and employs a masked autoencoder to reconstruct the original data, learning robust and informative cell representations. scMAE effectively captures latent structures and dependencies in the data, enhancing clustering performance.
scMAE employs partial corruption to the gene expression data and incorporates a masking predictor to capture the correlations between genes. scMAE takes the corrupted data as input to the encoder, obtains a low-dimensional embedding, and then passes it to the masking predictor.
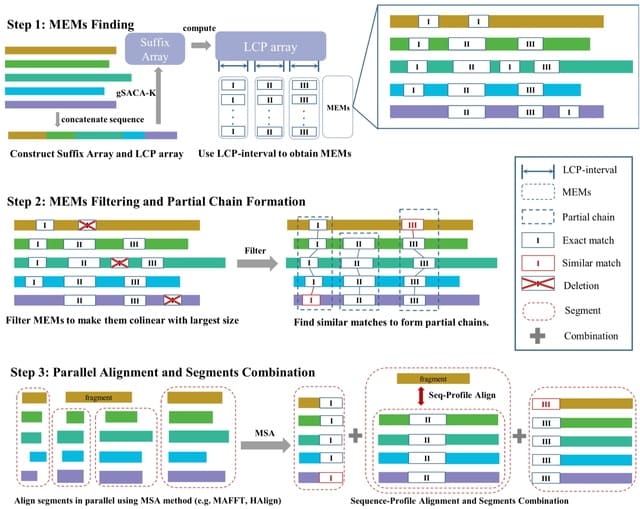
□ FMAlign2: a novel fast multiple nucleotide sequence alignment method for ultralong datasets
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae014/7515251
FMAlign2 utilizes Maximal Exact Matches (MEMs) instead of k-mers to identify partial chains in sequences. FMAlign2 constructs suffix array and longest common prefix (LCP) array, identifies MEMs, and generates a colinear set of MEMs for alignment.
FMAlign2 employs the striped Smith-Waterman (SSW) algorithm to identify similar substrings for each MEMs in sequences where MEMs are absent. The identified substrings, combined with MEMs, form the partial chains used for subsequent sequence segmentation to generate segments.
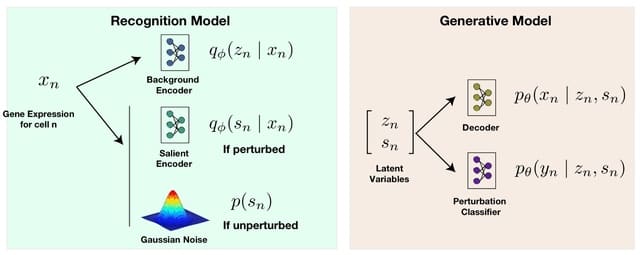
□ SC-VAE: A Supervised Contrastive Framework for Learning Disentangled Representations of Cell Perturbation Data
>> https://www.biorxiv.org/content/10.1101/2024.01.05.574421v1
SC-VAE (Supervised Contrastive Variational Autoencoder), a novel framework for learning disentangled representations from Perturb-Seq data. SC-VAE learns two latent spaces with the same semantic, but also jointly models guide RA identity alongside gene expression measurements.
SC-VAE employs the Hilbert-Schmidt Independence Criterion as a regularization technique. SC-VAE extends the CA framework by adding a supervision component to the generative model.
SC-VAE incorporates two distinct encoders: a background encoder, capturing biological attributes like cell cycle processes, and a salient encoder, specifically targeting perturbation effects.
The salient space induces a much higher energy distance compared to the background space, suggesting that the two spaces are disentangled. The energy distances for SC-VAE's salient space were consistently higher than those for ContrastiveVI's salient space or for the PCA space.
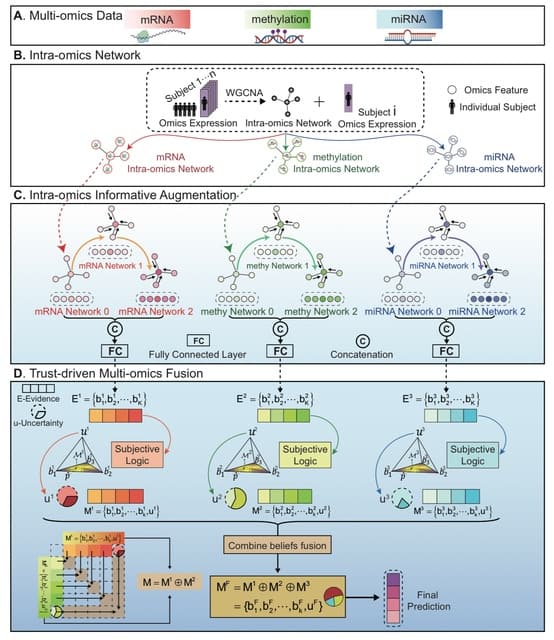
□ TEMINET: A Co-Informative and Trustworthy Multi-Omics Integration Network for Diagnostic Prediction
>> https://www.biorxiv.org/content/10.1101/2024.01.03.574118v1
TEMINET utilizes intra-omics features to construct disease-specific networks, then applies graph attention networks and a multi-level framework to capture more collective informativeness than pairwise relations.
TEMINET operates on a sample-wise basis with multi-omics information for each individual sample being imported into the model. The first intra-omics network is built using the WGCNA. The intra-omic information at each omics-level is augmented using the multi-level GAT.
The evidence is evaluated by the subject logic module to obtain uncertainty. During the integration phase, the trustworthy informativeness and uncertainty from each omics are amalgamated into a composite embedding encompassing inter-omics information.
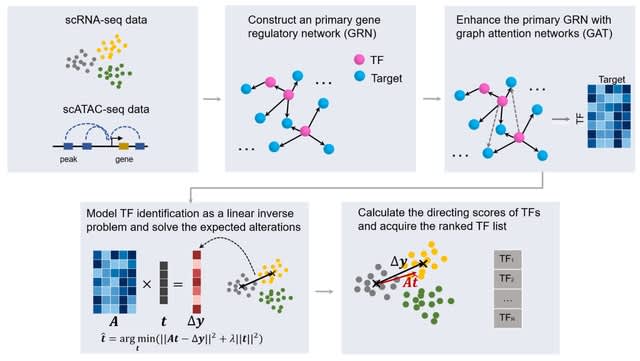
□ scDirect: key transcription factor identification for directing cell state transitions based on single-cell multi-omics data
>> https://www.biorxiv.org/content/10.1101/2024.01.08.574757v1
scDirect models cell state transition as a linear process. scDirect constructs a primary GRN with scRNA-seq data and scATAC-seq data, and then enhances the GRN with graph attention network (GAT) to obtain more putative TF-target pairs with high confidence.
scDirect uses CellOracle to calculate a primary GRN, and then GAT was applied to enhance the GRN. scDirect models the TF identification task as a linear inverse problem and solves the expected alteration of each TF with Tikhonov regularization.
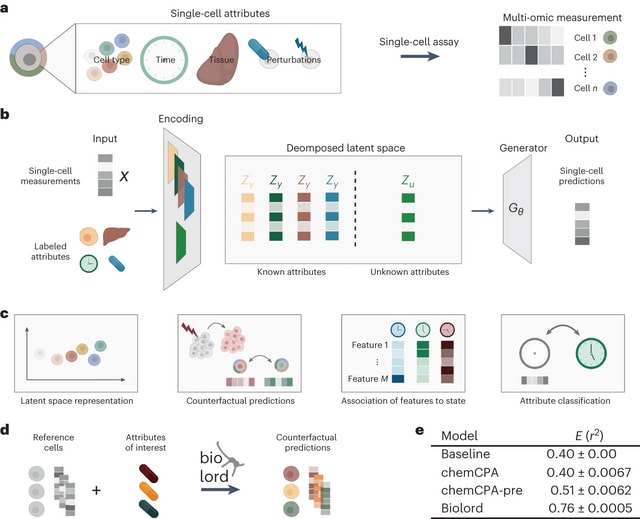
□ Biolord: Disentanglement of single-cell data
>> https://www.nature.com/articles/s41587-023-02079-x
Biolord is a deep generative method for disentangling single-cell multi-omic data to known and unknown attributes, including spatial, temporal and disease states, used to reveal the decoupled biological signatures over diverse single-cell modalities and biological systems.
Decomposed latent space - for each known attribute, a dedicated subnetwork is constructed. The architecture of each subnetwork is chosen based on the attributes' type (categorical or ordered),
The decomposed latent space and the generative prediction, is done jointly, such that the embeddings in the decomposed latent space are optimized with respect to the reconstruction error of the generator.
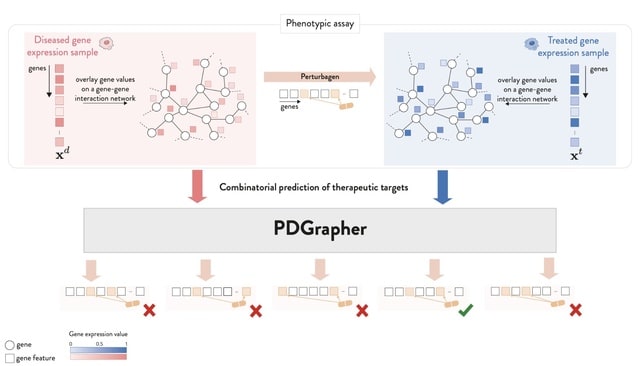
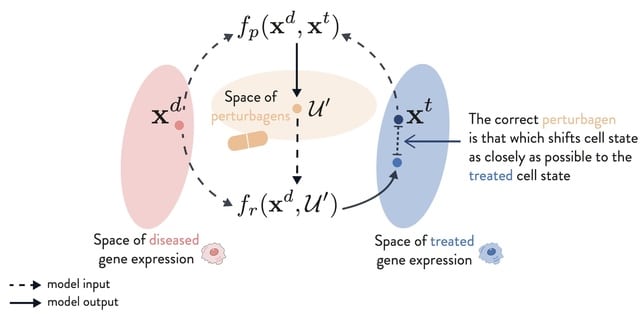
□ PDGrapher: Combinatorial prediction of therapeutic perturbations using causally-inspired neural networks
>> https://www.biorxiv.org/content/10.1101/2024.01.03.573985v2
PDGRAPHER efficiently predicts perturbagens to shift cell line gene expression from a diseased to a treated state across two evaluation settings and eight datasets of genetic and chemical interventions.
Training PDGRAPHER models is up to 30 times faster than response prediction methods that use indirect prediction to nominate candidate perturbagens.
PDGRAPHER can illuminate the mode of action of predicted perturbagens given that it predicts gene targets based on network proximity which governs similarity between genes.
PDGRAPHER posits that leveraging representation learning can overcome incomplete causal graph approximations. A valuable research direction is to theoretically examine the impact of using the approximations, focusing on how they influence the reliability of predicted likelihoods.
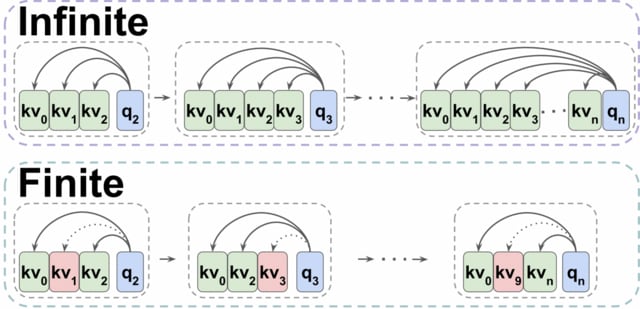
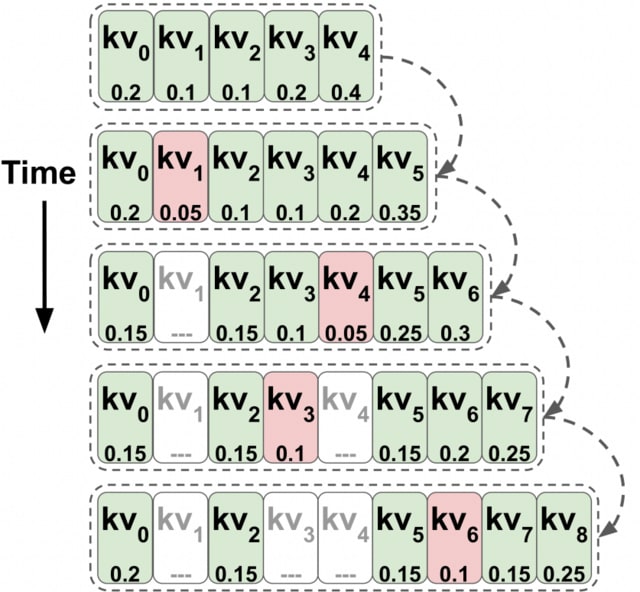
□ Transformers are Multi-State RNNs
>> https://arxiv.org/abs/2401.06104
Transformers can be thought of as infinite multi-state RNNs, with the key/value vectors corresponding to a multi-state that dynamically grows infinitely. Transformers behave as finite MSRNNs, which keep a fixed-size multi-state by dropping one state at each decoding step.
TOVA is a powerful MSRNN compression policy. TOVA selects which tokens to keep in the multi-state based solely on their attention scores. TOVA performs comparably to the infinite MSRNN model. Although transformers are not trained as such, they often function as finite MSRNNs.
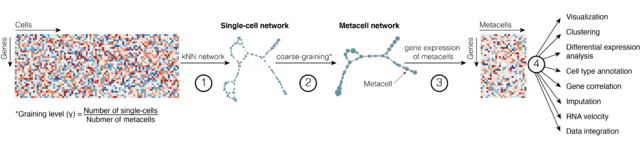
□ SuperCell: Coarse-graining of large single-cell RNA-seq data into metacells
>> https://github.com/GfellerLab/SuperCell
SuperCell is an R package for coarse-graining large single-cell RNA-seq data into metacells and performing downstream analysis at the metacell level.
Unlike clustering, the aim of metacells is not to identify large groups of cells that comprehensively capture biological concepts, like cell types, but to merge cells that share highly similar profiles, and may carry repetitive information.
Therefore metacells represent a compromise structure that optimally remove redundant information in scRNA-seq data while preserving the biologically relevant heterogeneity.
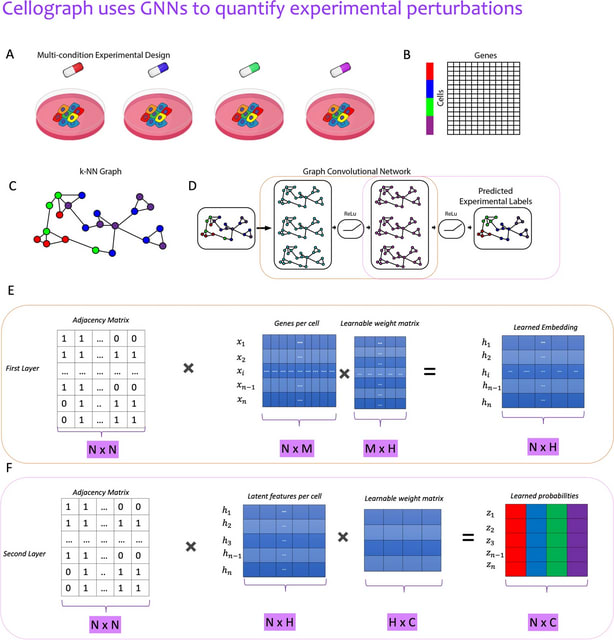
□ Cellograph: a semi-supervised approach to analyzing multi-condition single-cell RNA-sequencing data using graph neural networks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05641-9
Cellograph uses Graph Convolutional Networks (GCNs) to perform node classification on cells from multiple samples to quantify how representative cells are of each sample.
Cellograph not only measures how prototypical cells are of each condition but also learns a latent space that is amenable to interpretable data visualization and clustering. The learned gene weight matrix from training reveals pertinent genes driving the differences between conditions.
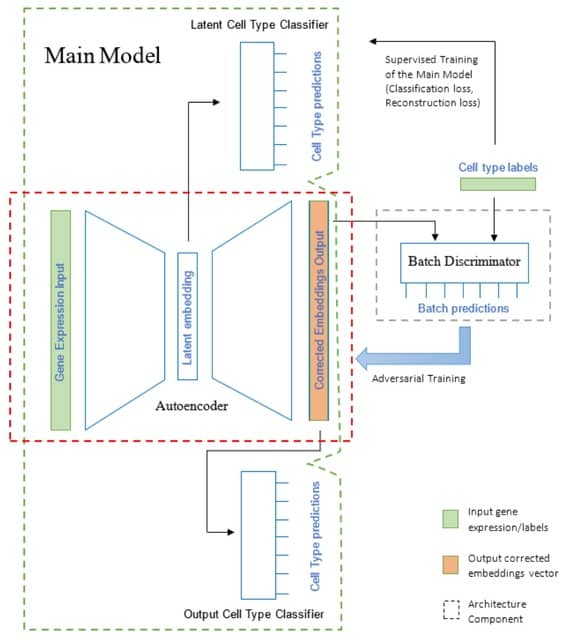
□ ABC: Batch correction of single cell sequencing data via an autoencoder architecture
>> https://academic.oup.com/bioinformaticsadvances/advance-article/doi/10.1093/bioadv/vbad186/7502962
Autoencoder-based Batch Correction (ABC), a semi-supervised deep learning architecture for integrating single cell sequencing. ABC removes batch effects through a guided process of data compression using supervised cell type classifier branches for biological signal retention.
ABC is based on an autoencoder architecture trained in an adversarial manner alongside a batch label discriminator, similar to GANs.
The architecture takes as input molecular measurements from a given cell, containing the normalized counts of each locus/gene in the cell, and outputs a corrected vector of values that can be used for downstream analysis.
In ABC approach, cell type classifiers are utilized to guide both encoding and decoding processes, ensuring the retention of cell type-specific variations. This is particularly relevant for cell types that are unique to a specific batch and represented by a small number of cells.
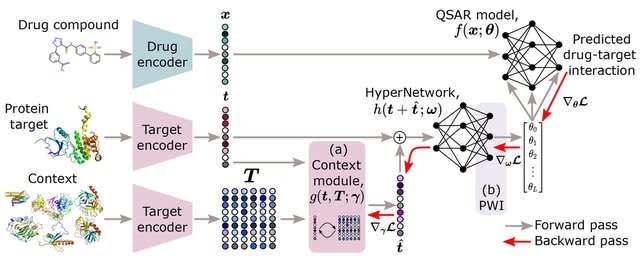
□ HyperPCM: Robust Task-Conditioned Modeling of Drug–Target Interactions
>> https://pubs.acs.org/doi/10.1021/acs.jcim.3c01417
HyperPCM, a novel neural network architecture that achieves state-of-the-art performance in various settings including during zero-shot inference, where predictions are made for previously unseen protein targets.
HyperPCM leverages the power of a HyperNetwork that learn to predict parameters for other neural networks. The specialized weight initialization strategy of the HyperNetwork stabilizes the signal propagation through the QSAR model.
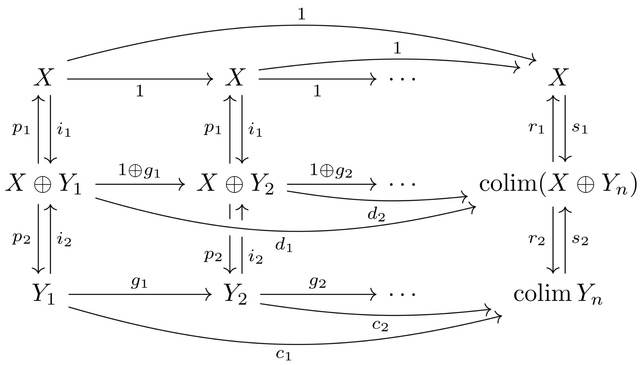
□ Dagger categories and the complex numbers: Axioms for the category of finite-dimensional Hilbert spaces and linear contractions
>>
Characterising the category of finite-dimensional Hilbert spaces and linear contractions using simple category-theoretic axioms that do not refer to norms, continuity, dimension, or real numbers.
The scalar localisation of a category satisfying this axioms is equivalent to the category of finite-dimensional Hilbert spaces and all linear maps, then identify the original category with the full subcategory of linear contractions.
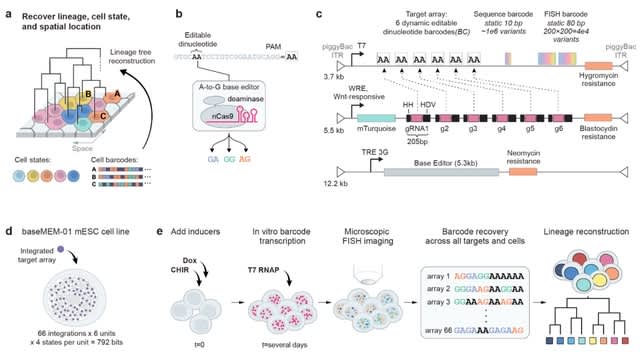
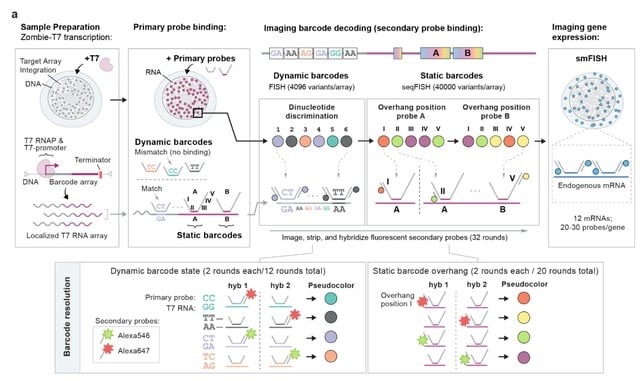
□ BaseMEMOIR: Reconstructing cell histories in space with image-readable base editor recording
>> https://www.biorxiv.org/content/10.1101/2024.01.03.573434v1
baseMEMOIR combines base editing, sequential hybridization imaging, and Bayesian inference to allow reconstruction of high-resolution cell lineage trees and cell state dynamics while preserving spatial organization.
BaseMEMOIR stochastically and irreversibly edits engineered dinucleotides to one of three alternative image-readable states. baseMEMOIR achieves high density recording, while maintaining compatibility with FISH-based readout of endogenous genes.
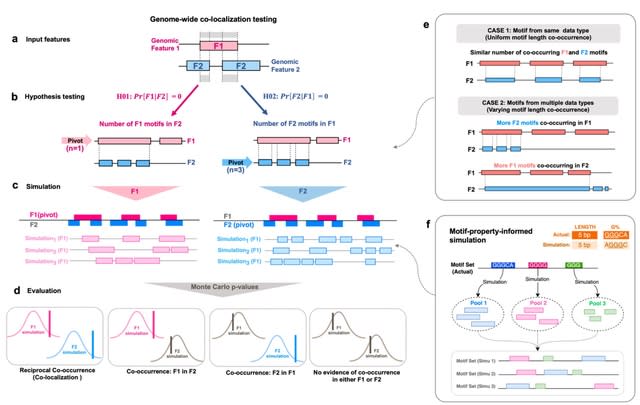
□ MoCoLo: a testing framework for motif co-localization
>> https://www.biorxiv.org/content/10.1101/2024.01.04.574249v1
MoCoLo employs a unique approach to co-localization testing that directly probes for genomic co-localization with duo-hypotheses testing. This means that MoCoLo can deliver more detailed and nuanced insights into the interplay between different genomic features.
MoCoLo features a novel method for informed genomic simulation, taking into account intrinsic sequence properties such as length and guanine-content.
MoCoLo enables us to identify genome-wide co-localization of 8-oxo-dG sites and non-B DNA forming region, providing a deeper understanding of the interactions between these genomic elements.
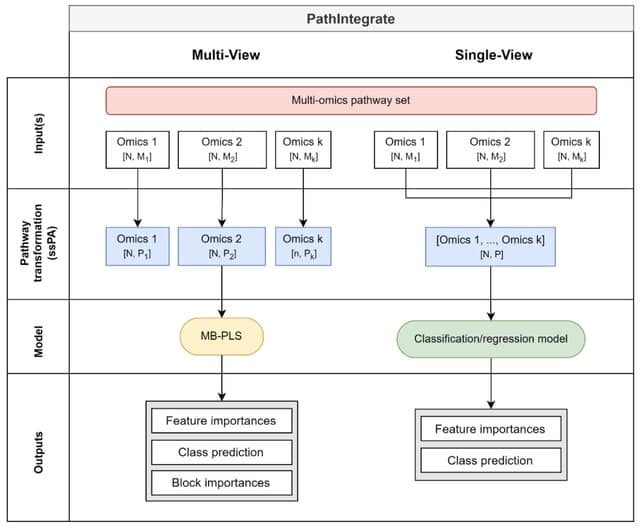
□ PathIntegrate: Multivariate modelling approaches for pathway-based multi-omics data integration
>> https://www.biorxiv.org/content/10.1101/2024.01.09.574780v1
PathIntegrate employs single-sample pathway analysis (ssPA) to transform multi-omics datasets from the molecular to the pathway-level, and applies a predictive single-view or multi-view model to integrate the data.
PathIntegrate Single-View produces a multi-omics pathway-transformed dataset and applies a classification or regression model. PathIntegrate Multi-View uses a multi-block partial least squares (MB-PLS) latent variable model to integrate ssPA-transformed multi-omics data.
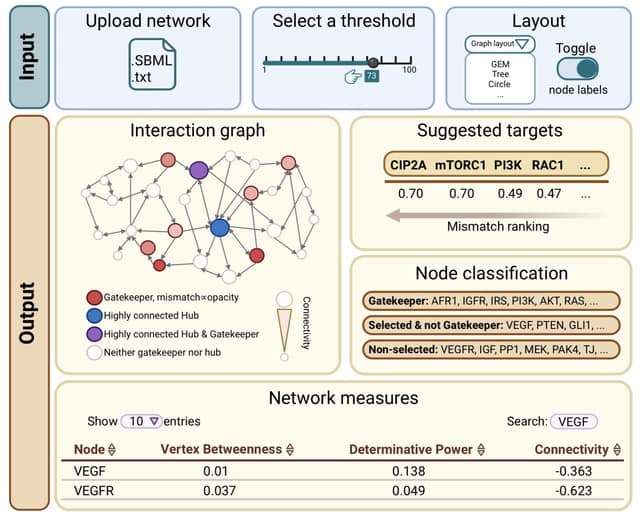
□ GatekeepR: an R shiny application for the identification of nodes with high dynamic impact in boolean networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae007/7513690
GatekeepR provides a ranked list of network components whose perturbation (i.e. knockout or overexpression) is likely to have a high impact on dynamics, resulting in a large change in the system's attractor landscape.
Such a change is defined by the loss of previously existing attractors along with the appearance of new attractors which possess a high Hamming distance with respect to all attractors of the unperturbed system.
The recommended nodes have been found to be sparsely connected and to preferentially exchange mutual information with highly connected hub nodes and have thus been named "gatekeepers".
GatekeepR does not perform any analyses on the state transition graph of a network, which scales exponentially with network size, but relies only on measures defined by the network's logical rules and their resulting interaction graph.
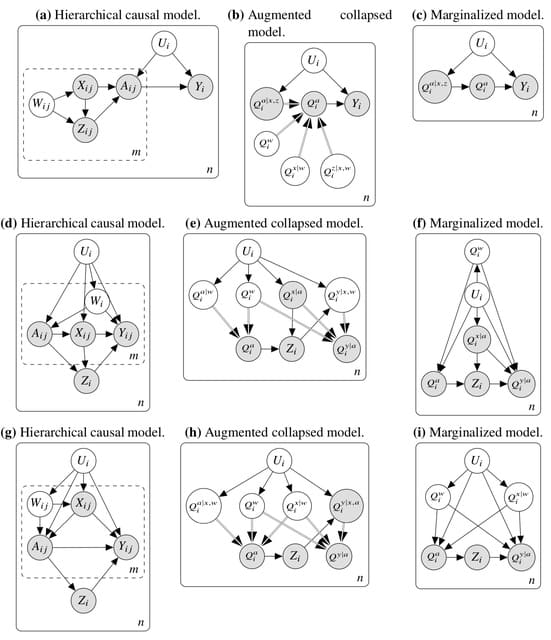
□ Hierarchical Causal Models
>> https://arxiv.org/abs/2401.05330
Hierarchical causal models (HCM), which extend structural causal models and causal graphical models by adding inner plates. It uses a general graphical identification technique for hierarchical causal models that extends do-calculus.
In the HCM identification problem, Infinite data from both units and subunits is considered. We find many situations in which hierarchical data can enable causal identification even when it would be impossible with non-hierarchical data.
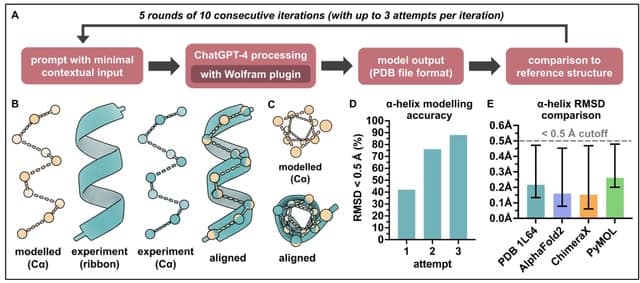
□ Generative artificial intelligence performs rudimentary structural biology modelling
>> https://www.biorxiv.org/content/10.1101/2024.01.10.575113v1
Using ChatGPT to model 3D structures for the 20 standard amino acids as well as an a-helical polypeptide chain, with the latter involving incorporation of the Wolfram plugin for advanced mathematical computation.
For amino acid modelling, distances and angles between atoms of the generated structures in most cases approximated to around experimentally-determined values.
For a-helix modelling, the generated structures were comparable to that of an experimentally-determined a-helical structure. However, both amino acid and a-helix modelling were sporadically error-prone and increased molecular complexity was not well tolerated.
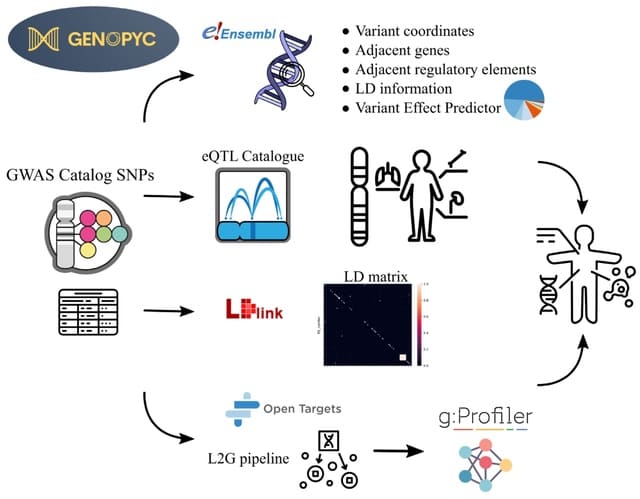
□ Genopyc: a python library for investigating the genomic basis of complex diseases
>> https://www.biorxiv.org/content/10.1101/2024.01.11.575316v1
Genopyc performs various tasks such as retrieve the functional elements neighbouring genomic coordinates, annotate variants, retrieving genes affected by non coding variants and perform and visualize functional enrichment analysis.
Genopyc can also retrieve a linkage-disequilibrium (LD) matrix for a set of SNPs by using LDlink, converting genome coordinates between genome versions and retrieving genes coordinates in the genome.
Genopyc queries the variant effect predictor (VEP) to predict the consequences of the SNPs on the transcript and its effect on neighboring genes and functional elements.
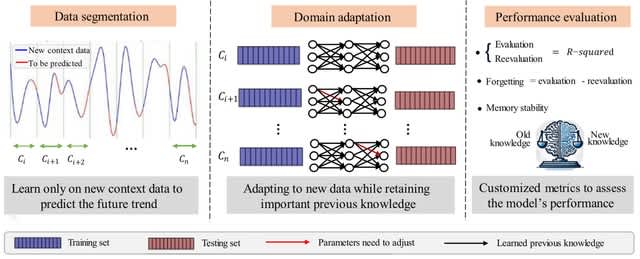
□ CEL: A Continual Learning Model for Disease Outbreak Prediction by Leveraging Domain Adaptation via Elastic Weight Consolidation
>> https://www.biorxiv.org/content/10.1101/2024.01.13.575497v1
CEL (Continual Learning by EWC and LSTM), a model for disease outbreak prediction designed to combat catastrophic forgetting in domain-incremental learning setting where the Fisher Information Matrix in Elastic Weight Consolidation is used to construct a regularization term.
CEL starts w/ data segmentation for contextual learning, followed by domain adaptation where a neural network incorporates with EWC and retains earlier knowledge while integrating new contexts. Finally, performance evaluation measures knowledge retention versus new learning.
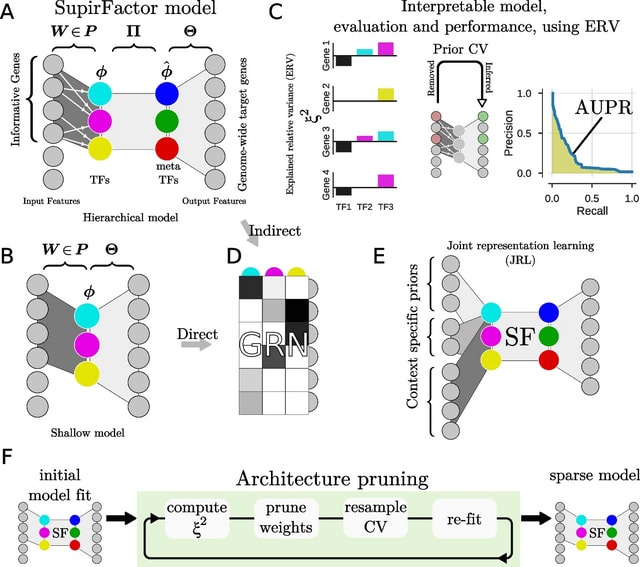
□ SupirFactor: Structure-primed embedding on the transcription factor manifold enables transparent model architectures for gene regulatory network and latent activity inference
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03134-1
SupirFactor (StrUcture Primed Inference of Regulation using latent Factor ACTivity), a novel autoencoder-based framework for modeling, and a metric, explained relative variance (ERV), for interpretation of GRNs.
SupirFactor incorporates knowledge priming by using prior, known regulatory evidence to constrain connectivity between an input gene expression layer and the first latent layer, which is explicitly defined to be TF-specific.