









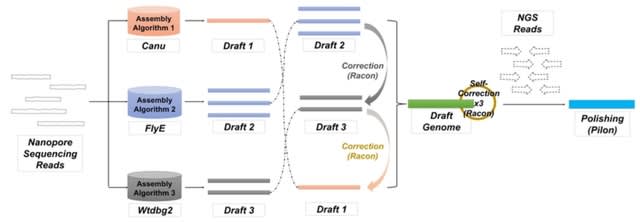
□ MAECI: A Pipeline For Generating Consensus Sequence With Nanopore Sequencing Long-read Assembly and Error Correction
>> https://www.biorxiv.org/content/10.1101/2022.04.04.487014v1.full.pdf
The assemblies can be corrected using nanopore sequencing data and then polished with NGS data. Both approaches can mitigate some of these problems and improve the accuracy of the assemblies, but assembly errors cannot be completely avoided.
MAECI enables the assembly for nanopore long-read sequencing data. It takes nanopore sequencing data as input, uses multiple assembly algorithms to generate a single consensus sequence, and then uses nanopore sequencing data to perform self-error correction.
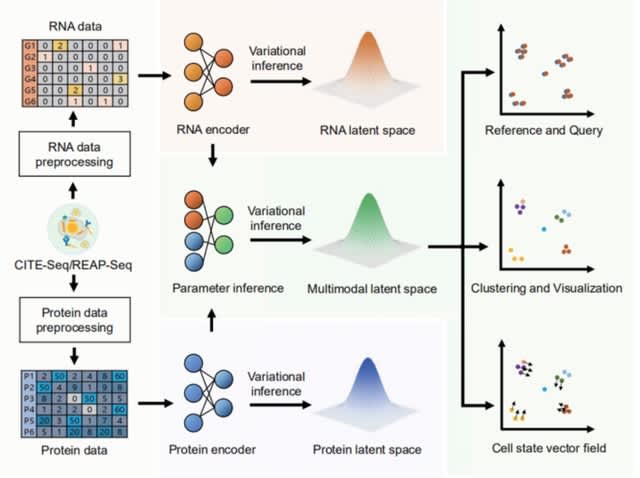
□ DPI: Single-cell multimodal modeling with deep parametric inference
>> https://www.biorxiv.org/content/10.1101/2022.04.04.486878v1.full.pdf
DPI, a deep parameter inference model that integrates CITE-seq/REAP-seq data. With DPI, the cellular heterogeneity embedded in the single-cell multimodal omics can be comprehensively understood from multiple views.
DPI describes the state of all cells in the sample in terms of the multimodal latent space. The multimodal latent space generated by DPI is continuous, which means that perturbing the genes/proteins of cells in the sample can find the cell state closest to it in this space.
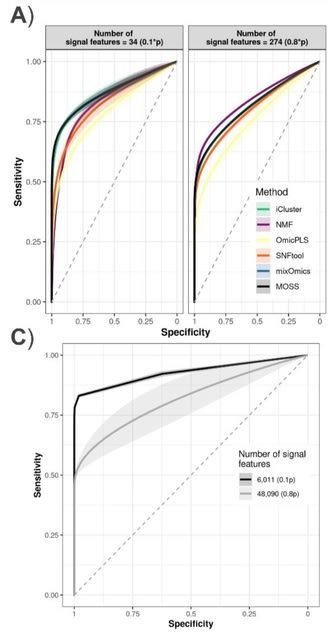
□ MOSS: Multi-omic integration with Sparse Value Decomposition
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac179/6553658
MOSS performs a Sparse Singular Value Decomposition (sSVD) on the integrated omic blocks to obtain latent dimensions as sparse factors (i.e., with zeroed out elements), representing variability across subjects and features.
MOSS can fit supervised analyses via partial least squares, linear discriminant analysis, and low-rank regressions. Sparsity is imposed via Elastic Net on the sSVD solutions. MOSS allows an automatic tuning of the number of elements different from zero.

□ GPS-seq: The DNA-based global positioning system—a theoretical framework for large-scale spatial genomics
>> https://www.biorxiv.org/content/10.1101/2022.03.22.485380v1.full.pdf
GPS-seq, a theoretical framework that enables massively scalable, optics-free spatial transcriptomics. GPS-seq combines data from high-throughput sequencing with manifold learning to obtain the spatial transcriptomic landscape of a given tissue section without optical microscopy.
In this framework, similar to technologies like Slide-seq and 10X Visium, tissue samples are stamped on a surface of randomly-distributed DNA-barcoded spots (or beads). The transcriptomic sequences of proximal cells are fused to DNA barcodes.
The barcode spots serve as “anchors” which also capture spatially diffused “satellite” barcodes, and therefore allow computational reconstruction of spot positions without optical sequencing or depositing barcodes to pre-specified positions.
The general framework of GPS-seq is also compatible with standard single-cell (or single-nucleus) capture methods, and any modality of single- cell genomics, such as sci-ATAC-seq, could be transformed into spatial genomics in this strategy.
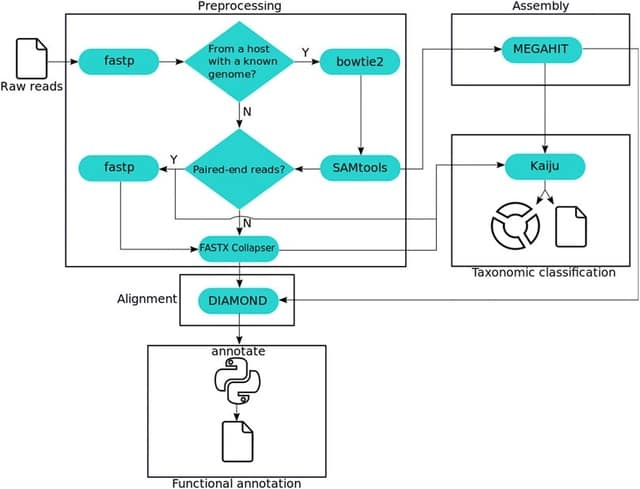
□ MEDUSA: A Pipeline for Sensitive Taxonomic Classification and Flexible Functional Annotation of Metagenomic Shotgun Sequences
>> https://www.frontiersin.org/articles/10.3389/fgene.2022.814437/full
MEDUSA performs preprocessing, assembly, alignment, taxonomic classification, and functional annotation on shotgun data, supporting user-built dictionaries to transfer annotations to any functional identifier.
MEDUSA includes several tools, as fastp, Bowtie2, DIAMOND, Kaiju, MEGAHIT, and a novel tool implemented in Python to transfer annotations to BLAST/DIAMOND alignment results.
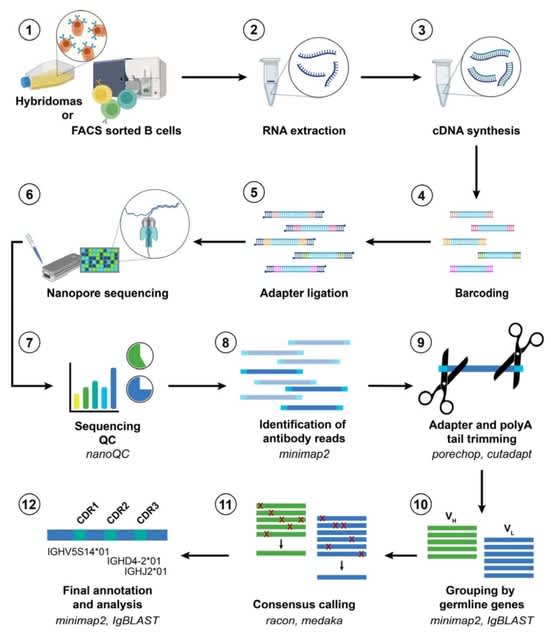
□ NAb-seq: an accurate, rapid and cost-effective method for antibody long-read sequencing in hybridoma cell lines and single B cells
>> https://www.biorxiv.org/content/10.1101/2022.03.25.485728v1.full.pdf
When compared to Sanger sequencing of two hybridoma cell lines, long-read ONT sequencing was highly accurate, reliable, and amenable to high throughput.
NAb-seq, a three-day, species-independent, and cost-effective workflow to characterize paired full- length immunoglobulin light and heavy chain genes from hybridoma cell lines.

□ SimSCSnTree: a simulator of single-cell DNA sequencing data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac169/6551250
SimSCSnTree, a new single-cell DNA sequence simulator which generates an evolutionary tree of cells and evolves single nucleotide variants (SNVs) and copy number aberrations (CNAs) along its branches.
Data generated by the simulator can be used to benchmark tools for single-cell genomic analyses, particularly in cancer where SNVs and CNAs are ubiquitous.
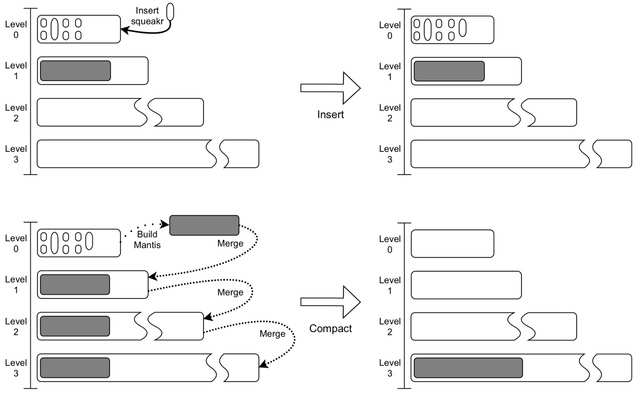
□ Dynamic Mantis: An Incrementally Updatable and Scalable System for Large-Scale Sequence Search using the Bentley-Saxe Transformation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac142/6553005
an efficient algorithm for merging two Mantis indexes, and tackle several scalability and efficiency obstacles along the way. The proposed algorithm targets Minimum Spanning Tree-based Mantis.
MST-based Mantis is ≈ 10× faster to construct, requires ≈ 10× less construction memory, results in ≈ 2.5× smaller indexes, and performs bulk queries ≈ 74× faster and with ≈ 100× less query memory than Bifrost.

□ Triku: a feature selection method based on nearest neighbors for single-cell data
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giac017/6547682
Triku is a feature selection method that favors genes defining the main cell populations. It does so by selecting GE by groups of cells that are close in the k-NN graph. The expression of these genes is higher than the expected expression if the k-cells were chosen at random.
the Wasserstein distance between the observed and the expected distributions is computed and genes are ranked according to that distance. Higher distances imply that the gene is locally expressed in a subset of transcriptomically similar cells.

□ RF4Del: A Random Forest approach for accurate deletion detection
>> https://www.biorxiv.org/content/10.1101/2022.03.10.483419v1.full.pdf
The model consists of 13 features extracted from a mapping file. RF4Del outperforms established SV callers (DELLY, Pindel) with higher overall performance (F1-score > 0.75; 6x-12x sequence coverage) and is less affected by low sequencing coverage and deletion size variations.
RF4Del could learn from a compilation of sequence patterns linked to a given SV. Such models can then be combined to form a learning system able to detect all types of SVs in a given genome.

□ GRAPE: Genomic Relatedness Detection Pipeline
>> https://www.biorxiv.org/content/10.1101/2022.03.11.483988v1.full.pdf
GRAPE: Genomic RelAtedness detection PipelinE. It combines data preprocess- ing, identity-by-descent (IBD) segments detection, and accurate relationship esti- mation.
GRAPE has a modular architecture that allows switching between tools and adjust tools parameters for better control of precision and recall levels. The pipeline also contains a simulation workflow w/ an in-depth evaluation of pipeline accuracy using simulated and reference data.
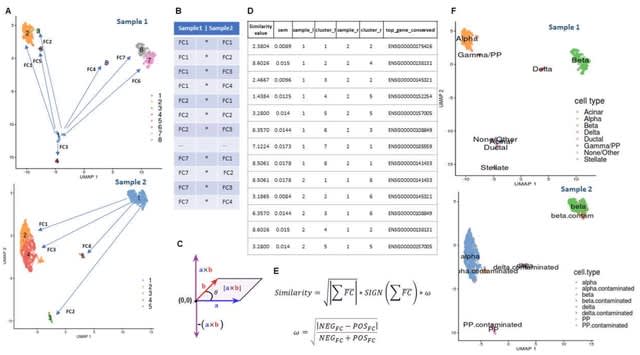
□ ClusterFoldSimilarity: A single-cell clusters similarity measure for different batches, datasets, and samples
>> https://www.biorxiv.org/content/10.1101/2022.03.14.483731v1.full.pdf
ClusterFoldSimilarity calculates a measure of similarity b/n clusters from different datasets/batches, without the need of correcting for batch effect or normalizing and merging the data, thus avoiding artifacts and the loss of information derived from these kinds of techniques.
The similarity metric is based on the average vector module and sign of the product of logarithmic fold-changes. ClusterFoldSimilarity compares every single pair of clusters from any number of different samples/datasets, including different number of clusters for each sample.
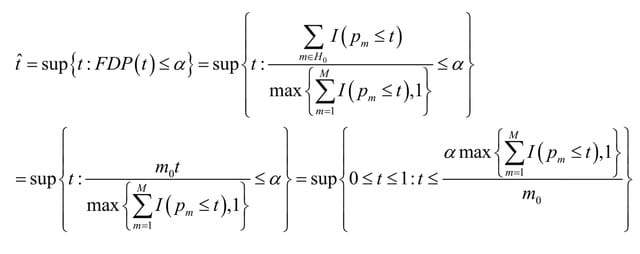
□ HCLC-FC: a novel statistical method for phenome-wide association studies
>> https://www.biorxiv.org/content/10.1101/2022.03.14.484203v1.full.pdf
HCLC-FC (Hierarchical Clustering Linear Combination with False discovery rate Control), to test the association between a genetic variant with multiple phenotypes for each phenotypic category in phenome-wide association studies (PheWAS).
HCLC-FC clusters phenotypes within each phenotypic category, which reduces the degrees of freedom of the association tests and has the potential to increase statistical power. HCLC-FC has an asymptotic distribution which avoids the computational burden of simulation.
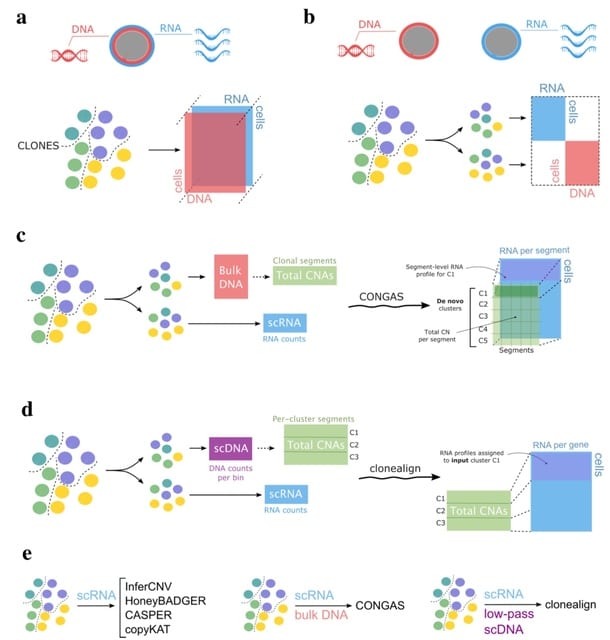
□ CONGAS: A Bayesian method to cluster single-cell RNA sequencing data using Copy Number Alterations
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac143/6550058
CONGAS jointly identifies clusters of single cells with subclonal copy number alterations, and differences in RNA expression.
CONGAS builds statistical priors leveraging bulk DNA sequencing data, does not require a normal reference and scales fast thanks to a GPU backend and variational inference.

□ OMAMO: orthology-based alternative model organism selection
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac163/6550503
The only unicellular organisms considered in these databases are fission and budding yeast, whilst abundance of unicellular species in nature and their unique features make it difficult to find other non-complex model organisms for a biological process of interest.
OMAMO (Orthologous Matrix and Alternative Model Organisms), a software and a web service that provide the user with the best non-complex organism for research into a biological process of interest based on orthologous relationships between human and the species.
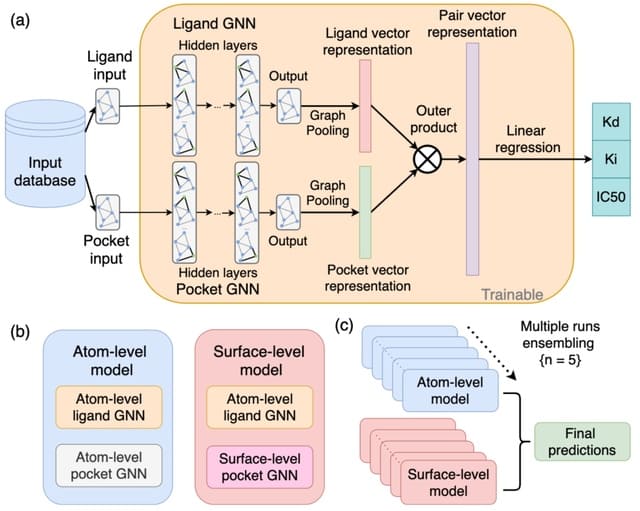
□ DENVIS: scalable and high-throughput virtual screening using graph neural networks with atomic and surface protein pocket features
>> https://www.biorxiv.org/content/10.1101/2022.03.17.484710v1.full.pdf
DENVIS, a purely machine learning-based, high-throughput, end-to-end-strategy for SBVS using GNNs for binding affinity prediction. DENVIS exhibits several orders of magnitude faster screening times (i.e., higher throughput) than both docking-based and hybrid models.
The atom-level model consists of a modified version of the graph isomorphism network (GIN). The surface-level approach utilises a mixture model network (MoNet), a specialised GNN with a convolution operation that respects the geometry of the input manifold.
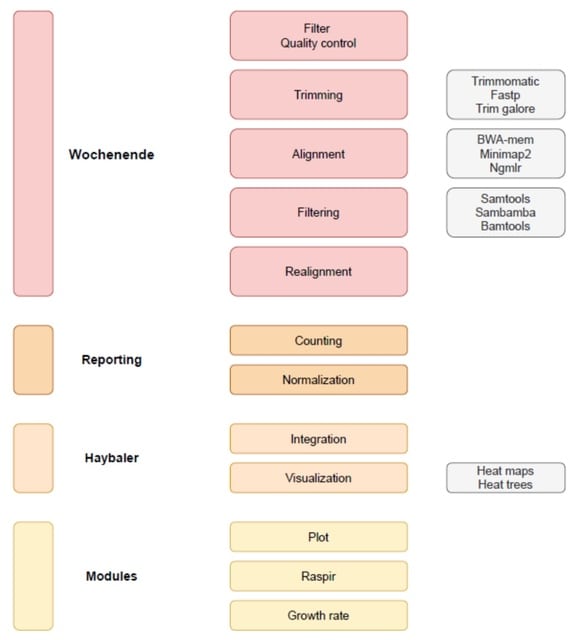
□ Wochenende - modular and flexible alignment-based shotgun metagenome analysis
>> https://www.biorxiv.org/content/10.1101/2022.03.18.484377v1.full.pdf
Wochenende runs alignment of short reads (eg Illumina) or long reads (eg Oxford Nanopore) against a reference sequence. It is relevant for genomics and metagenomics. Wochenende is simple (python script), portable and is easy to configure with a central config file.
Wochenende has the ability to find and filter alignments to all kingdoms of life using both short and long reads with high sensitivity and specificity, and provides the user with multiple normalization techniques and configurable and transparent filtering steps.

□ GBScleanR: Robust genotyping error correction using hidden Markov model with error pattern recognition.
>> https://www.biorxiv.org/content/10.1101/2022.03.18.484886v1.full.pdf
GBScleanR implements a novel HMM-based error correction algorithm. This algorithm estimates the allele read bias and mismap rate per marker and incorporates these into the HMM as parameters to capture the skewed probabilities in read acquisitions.
GBScleanR provides functions for data visualization, filtering, and loading/writing a VCF file. The algorithm of GBScleanR is based on the HMM and treats the observed allele read counts for each SNP marker along a chromosome as outputs from a sequence of latent true genotypes.

□ 3GOLD: optimized Levenshtein distance for clustering third-generation sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04637-7
3GOLD offers a novel way of determining error type and frequency by interpreting the unweighted SLD value and position on the matrix by comparing it to the unweighted LD value. 3GOLD combines the discriminatory benefits of weighted LD and the permissive benefits of SLD.
This approach is appropriate for datasets of unknown cluster centroids, such as those generated with unique molecular identifiers as well as known centroids such as barcoded datasets. It has high accuracy in resolving small clusters and mitigating the number of singletons.
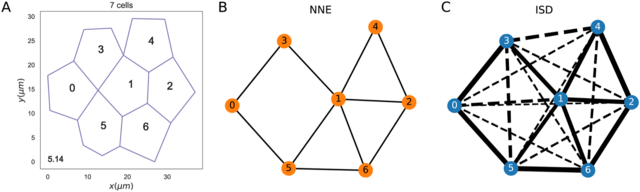
□ The role of cell geometry and cell-cell communication in gradient sensing
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009552
Generalizing the existing mathematical models to investigate how short- and long-range cellular communication can increase gradient sensing in two-dimensional models of epithelial tissues.
With long-range communication, the gradient sensing ability improves for tissues with more disordered geometries; on the other hand, an ordered structure with mostly hexagonal cells is advantageous with nearest neighbour communication.
□ Crimp: fast and scalable cluster relabeling based on impurity minimization
>> https://www.biorxiv.org/content/10.1101/2022.03.22.485309v1.full.pdf
CRIMP, a lightweight command-line tool, which offers a relatively fast and scalable heuristic to align clusters across multiple replicate clusterings consisting of the same number of clusters.
CRIMP allows to rearrange a number of membership matrices of identical shape in order to minimize differences caused by label switching. The remaining differences should be attributable to either noise or truly different ways of the data, referred to as ‘genuine multimodality’.
□ RabbitV: fast detection of viruses and microorganisms in sequencing data on multi-core architectures
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac187/6554196
RabbitV, a tool for rapid detection of viruses and microorganisms in Illumina sequencing datasets based on fast identification of unique k-mers. It can exploit the power of modern multi-core CPUs by using multi-threading, vectorization, and fast data parsing.
RabbitV outperforms fastv by a factor of at least 42.5 and 14.4 in unique k-mer generation (RabbitUniq) and pathogen identification (RabbitV), respectively.

□ q2-fondue: Reproducible acquisition, management, and meta-analysis of nucleotide sequence (meta)data
>> https://www.biorxiv.org/content/10.1101/2022.03.22.485322v1.full.pdf
q2-fondue (Functions for reproducibly Obtaining and Normalizing Data re-Used from Elsewhere) to expedite the initial acquisition of data from the SRA, while offering complete provenance tracking.
q2-fondue simplifies retrieval of sequencing data and accompanying metadata in a validated and standardized format interoperable with the QIIME 2 ecosystem.

□ MASI: Fast model-free standardization and integration of single-cell transcriptomics data
>> https://www.biorxiv.org/content/10.1101/2022.03.28.486110v1.full.pdf
MASI (Marker-Assisted Standardization and Integration) can run integrative annotation on a personal laptop for approximately one million cells, providing a cheap computational alternative for the single-cell data analysis community.
MASI will not be able to annotate cell types in query data that have not been seen in reference data.
However, it is still worth answering if a cell-type score matrix constructed using the reference data can preserve cell-type structure for query data, even though query data contains unseen cell types.
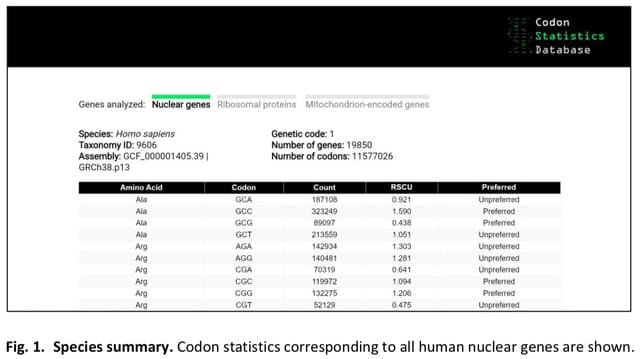
□ The Codon Statistics Database: a Database of Codon Usage Bias
>> https://www.biorxiv.org/content/10.1101/2022.03.29.486291v1.full.pdf
the Codon Statistics Database, an online database that contains codon usage statistics for all the species with reference or representative genomes in RefSeq.
If a species is selected, the user is directed to a table that lists, for each codon, the encoded amino acid, the total count in the genome, the RSCU, and whether the codon is preferred or unpreferred.

□ Boquila: NGS read simulator to eliminate read nucleotide bias in sequence analysis
>> https://www.biorxiv.org/content/10.1101/2022.03.29.486262v1.full.pdf
Boquila generates sequences that mimic the nucleotide profile of true reads, which can be used to correct the nucleotide-based bias of genome-wide distribution of NGS reads.
Boquila can be configured to generate reads from only specified regions of the reference genome. It also allows the use of input DNA sequencing to correct the bias due to the copy number variations in the genome.
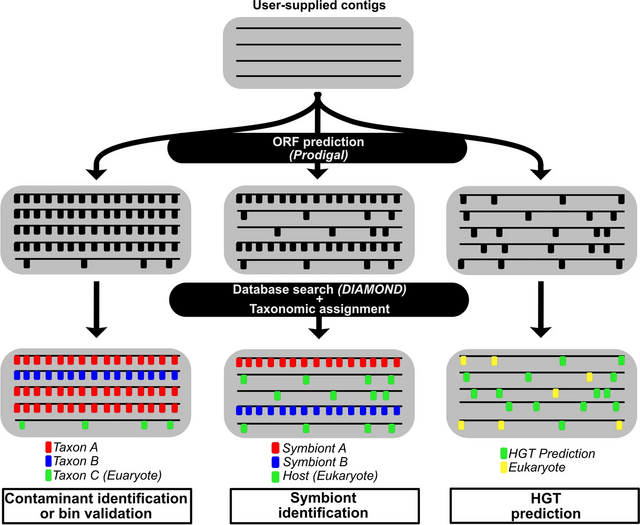
□ SprayNPray: user-friendly taxonomic profiling of genome and metagenome contigs
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08382-2
SprayNPray offers a quick and user-friendly, semi-automated approach, allowing users to separate contigs by taxonomy of interest. SprayNPray can be used for broad-level overviews, preliminary analyses, or as a supplement to other taxonomic classification or binning software.
SprayNPray profiles contigs using multiple metrics, including closest homologs from a user-specified reference database, gene density, read coverage, GC content, tetranucleotide frequency, and codon-usage bias.
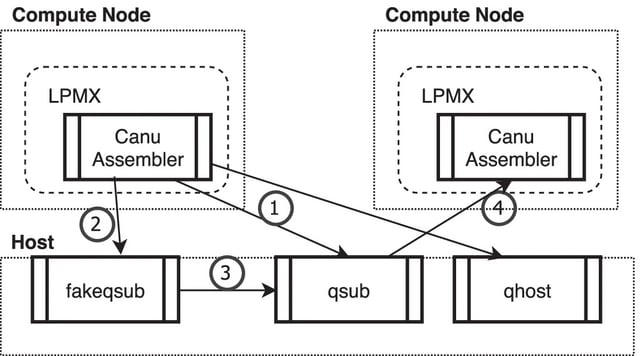
□ LPMX: a pure rootless composable container system
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04649-3
LPMX accelerates science by letting researchers compose existing containers and containerize tools/pipelines that are difficult to package/containerize using Conda or Singularity, thereby saving researchers’ precious time.
LPMX can minimize the overhead of splitting a large pipeline into smaller containerized components or tools to avoid conflicts between the components.
A caveat is that compared to Singularity, the LPMX approach might put a larger burden on a central shared file system, so Singularity might scale well beyond a certain large number of nodes.
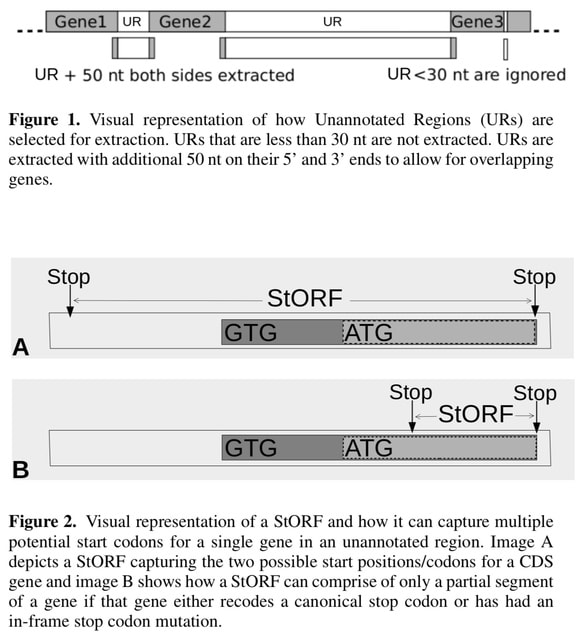
□ StORF-Reporter: Finding Genes between Genes
>> https://www.biorxiv.org/content/10.1101/2022.03.31.486628v1.full.pdf
StORF- Reporter, a tool that takes as input an annotated genome and returns missed CDS genes from the unannotated regions. Stop-ORFs (StORFs) are identified in these unannotated regions. StORFs are Open Reading Frames that are delimited by stop codons.
StORFs recovers complete coding sequences (with/without similarity to known genes) which were missing from both canonical and novel genome annotations.
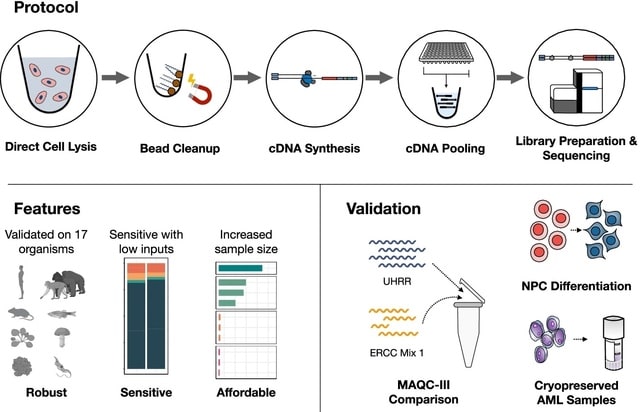
□ Prime-seq, efficient and powerful bulk RNA sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02660-8
Prime-seq, a bulk RNA-seq protocol, and show that it is as powerful and accurate as TruSeq in quantifying gene expression levels, but more sensitive and much more cost-efficient.
The prime-seq protocol is based on the SCRB-seq and the optimized derivative mcSCRB-seq. It uses the principles of poly(A) priming, template switching, early barcoding, and UMIs to generate 3′ tagged RNA-seq libraries.
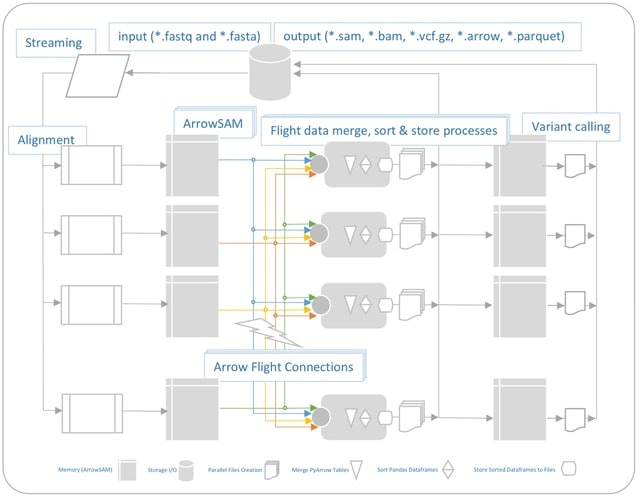
□ Communication-Efficient Cluster Scalable Genomics Data Processing Using Apache Arrow Flight
>> https://www.biorxiv.org/content/10.1101/2022.04.01.486780v1.full.pdf
This solution has similar performance to MPI-based HPC solutions, with the added advantage of easy programmability and transparent big data scalability. It outperforms existing Apache Spark based solutions in term of both computation time (2x) and lower communication overhead.
QUARTIC (QUick pArallel algoRithms for high-Throughput sequencIng data proCessing) is implemented using MPI. Though this implementation uses I/Os between pre-processing stages, it still performs better than other Apache Spark based frameworks.

□ epiAneufinder: identifying copy number variations from single-cell ATAC-seq data
>> https://www.biorxiv.org/content/10.1101/2022.04.03.485795v1.full.pdf
epiAneufinder, a novel algorithm that exploits the read count information from scATAC-seq data to extract genome-wide copy number variations (CNVs) for individual cells, allowing to explore the CNV heterogeneity present in a sample at the single-cell level.
epiAneufinder extracts single-cell copy number variations from scATAC-seq data alone, or alternatively from single-cell multiome data, without the need to supplement the data with other data modalities.
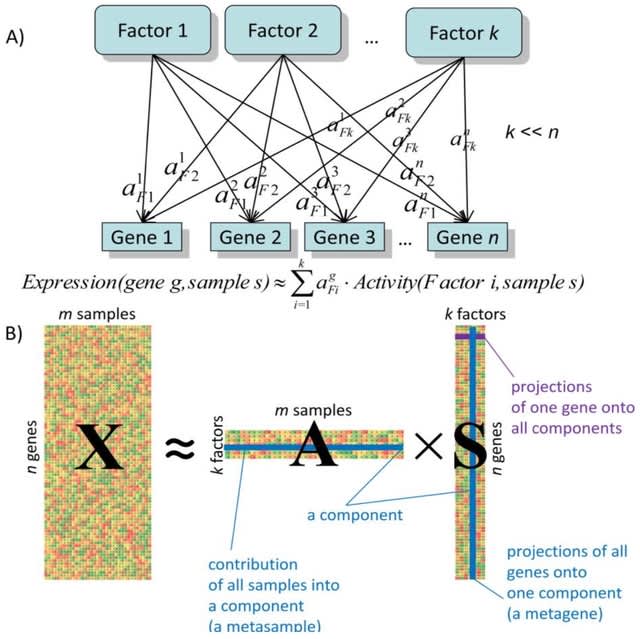
□ BIODICA: a computational environment for Independent Component Analysis
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac204/6564219
BIODICA, an integrated computational environment for application of Independent Component Analysis (ICA) to bulk and single-cell molecular profiles, interpretation of the results in terms of biological functions and correlation with metadata.
BIODICA automates deconvolution of large omics datasets with optimization of deconvolution parameters, and compares the results of deconvolution of independent datasets for distinguishing reproducible signals, universal and specific for a particular disease/data type or subtype.
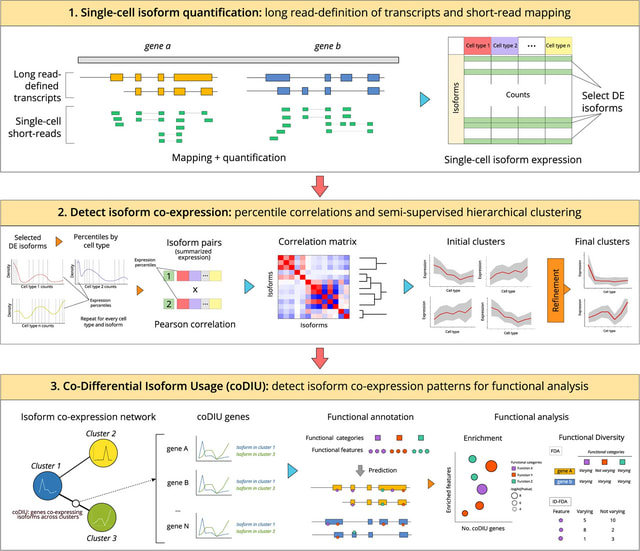
□ acorde unravels functionally interpretable networks of isoform co-usage from single cell data
>> https://www.nature.com/articles/s41467-022-29497-w
acorde, a pipeline that successfully leverages bulk long reads and single-cell data to confidently detect alternative isoform co-expression relationships.
acorde uses a strategy to obtain noise-robust correlation estimates in scRNA-seq data, and a semi-automated clustering approach to detect modules of co-expressed isoforms across cell types.
Percentile-summarized Pearson correlations outperform both classic and single-cell specific correlation strategies, including proportionality methods that were recently proposed as one of the best alternatives to measure co-expression in single-cell data.










※コメント投稿者のブログIDはブログ作成者のみに通知されます