










□ Chronumental: time tree estimation from very large phylogenies
>> https://www.biorxiv.org/content/10.1101/2021.10.27.465994v1.full.pdf
Chronumental uses stochastic gradient descent to identify lengths of time for tree branches which maximise the evidence lower bound under a probabilistic model, implemented in a framework which can be compiled into XLA for rapid computation.
Representing the summation of branch lengths to estimate node dates as a notional matrix multiplication, by constructing a vast matrix in which one dimension represents the leaf nodes, and one dimension represents the internal branches, with a 1 at each element.
When this matrix is multiplied by a vector of time-lengths for each branch it would yield the date corresponding to each leaf node. Such a matrix would contain over 10^12 elements, dwarfing any resources, but since almost all elements are 0s.
It can be represented as a “sparse matrix”, encoded in coordinate list (COO) format, with the matrix multiplication performed through ‘take’ and ‘segment_sum’ XLA operations.
Representing the operations in this way allows them to be efficiently compiled in XLA, which creates a differentiable graph of arithmetic operations.
Chronumental scales to phylogenies featuring millions of nodes, with chronological predictions made in minutes, and is able to accurately predict the dates of nodes for which it is not provided with metadata.

□ Stabilization of continuous-time Markov/semi-Markov jump linear systems via finite data-rate feedback
>> https://arxiv.org/pdf/2110.14931v1.pdf
the stabilization problem of the Markov jump linear systems (MJLSs) under the communication data-rate constraints, where the switching signal is a continuous-time Markov process. Sampling and quantization are used as fundamental tools to deal with the problem.
the sufficient conditions are given to ensure the almost sure exponential stabilization of the Markov jump linear systems. The conditions depend on the generator of the Markov process. The sampling times and the jump time is also independent.

□ Linear Approximate Pattern Matching Algorithm
>> https://www.biorxiv.org/content/10.1101/2021.10.25.465764v1.full.pdf
a structure that can be built in linear time and space and solve the approximate matching problem in (O(m + logΣk n/k! + occ) search costs, where m is the k! length of the pattern, n is the length of the reference, and k is the number of tolerated mismatches.
Building a novel index that index all suffixes under all internal nodes in the suffix tree in linear time and with maintaining the inter-connectivity among the suffixes under different internal nodes.
The non-linear time cost is due to the trivial process of checking whether each suffix under each internal node is already indexed in OT index. OSHR tree is constructed by reversing the suffix links in ST. Clearly, the space and time cost for building OSHR tree is linear (O(n)).

□ Counterfactuals in Branching Time: The Weakest Solution
>> https://arxiv.org/pdf/2110.11689v1.pdf
a formal analysis of temporally sensitive counterfactual condition- als, using the fusion of Ockhamist branching time temporal logic and minimal counterfactual logic P.
The main advantage of Ockhamist branching time theory in the context of counter- factuals is that it allows both expressions about time and historical possibility/necessity.
Atomic propositions and Boolean connectives have standard meaning. Gφ reads as ”at every moment in the future, φ”, Hφ – ”at every moment in the past, φ”, φ – ”it is historically necessary that φ”, which means that in all possible alternative histories, it is φ at the moment.

□ Model-free inference of unseen attractors: Reconstructing phase space features from a single noisy trajectory using reservoir computing
>> https://arxiv.org/pdf/2108.04074.pdf
A reservoir computer is able to learn the various attractors of a multistable system. In separate autonomous operation, the trained reservoir is able to reproduce and therefore infer the existence and shape of these unseen attractors.
the ability to learn the dynamics of a complex system can be extended to systems with multiple co-existing attractors, here a 4-dimensional extension of the well-known Lorenz chaotic system.
The reservoir computers are learning the phase space flow without formulating any intermediate model. They use a continuous time version of an echo state network based on ordinary differential equations.

□ Beyond sequencing: machine learning algorithms extract biology hidden in Nanopore signal data
>> https://www.cell.com/trends/genetics/fulltext/S0168-9525(21)00257-2
Nanopore sequencing accuracy has increased to 98.3% as new-generation base callers replace early generation hidden Markov model basecalling algorithms with neural network algorithms.
Nanopore direct RNA sequencing profiles RNAs with their modification retained, which influences the ion current signals emitted from the nanopore.
Machine learning and statistical testing tools can detect DNA modifications by analyzing ion current signals from nanopore direct DNA sequencing.
Machine learning methods can classify sequences in real-time, allowing targeted sequencing with nanopore’s ReadUntil feature.
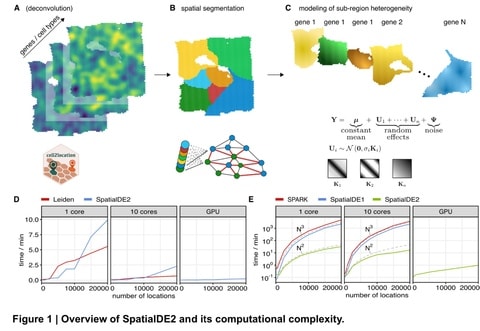
□ SpatialDE2: Fast and localized variance component analysis of spatial transcriptomics
>> https://www.biorxiv.org/content/10.1101/2021.10.27.466045v1.full.pdf
SpatialDE2 implements two major modules, which together provide for an end-to-end workflow for analyzing spatial transcriptomics data: a tissue region segmentation module and a module for detecting spatially variable genes.
SpatialDE2 provides a coherent model for tissue segmentation. Briefly, the spatial tissue region segmentation module is based on a Bayesian hidden markov random field, which segments tissues into distinct histological regions while explicitly accounting for spatial smoothness.

□ A Markov random field model for network-based differential expression analysis of single-cell RNA-seq data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04412-0
MRFscRNAseq is based on a Markov random field (MRF) model to appropriately accommodate gene network information as well as dependencies among cell types to identify cell-type specific DEGs.
With observed DE evidence, it utilizes a Markov random field model to appropriately take gene network information as well as dependencies among cell types into account.

□ SCA: Discovering Rare Cell Types through Information-based Dimensionality Reduction
>> https://www.biorxiv.org/content/10.1101/2021.01.19.427303v3.full.pdf
Shannon component analysis (SCA), a technique that leverages the information- theoretic notion of surprisal for dimensionality reduction. SCA’s information-theoretic paradigm opens the door to more meaningful signal extraction.
In cytotoxic T-cell data, SCA cleanly separates the gamma-delta and MAIT cell subpopulations, which are not detectable via PCA, ICA, scVI, or a wide array of specialized rare cell recovery tools.
SCA leverages the notion of surprisal, whereby less probable events are more informative when they occur, to assign an information score to each transcript in each cell.
□ MoNET: an R package for multi-omic network analysis
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab722/6409845
MoNET enables users to not only track down the interaction of SNPs/genes with metabolome level, but also trace back for the potential risk variants/regulators given altered genes/metabolites.
MoNET is expected to advance our understanding of the multi-omic findings by unveiling their trans-omic interactions and is likely to generate new hypotheses for further validation.
□ SigTools: Exploratory Visualization For Genomic Signals
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab742/6413626
Sigtools is an R-based visualization package, designed to enable the users with limited programming experience to produce statistical plots of continuous genomic data.
Sigtools consists of several statistical visualizations that provide insights regarding the behavior of a group of signals in large regions – such as a chromosome or the whole genome – as well as visualizing them around a specific point or short region.

□ Techniques to Produce and Evaluate Realistic Multivariate Synthetic Data
>> https://www.biorxiv.org/content/10.1101/2021.10.26.465952v1.full.pdf
The work demonstrates how to generate multivariate synthetic data that matches the real input data by converting the input into multiple one-dimensional (1D) problems.
The work also shows that it is possible to convert a multivariate input probability density function to a form that approximates a multivariate normal, although the technique is not dependent upon this finding.

□ RCX – an R package adapting the Cytoscape Exchange format for biological networks
>> https://www.biorxiv.org/content/10.1101/2021.10.26.466001v1.full.pdf
CX is a JSON-based data structure designed as a flexible model for transmitting networks with a focus on flexibility, modularity, and extensibility. Although those features are widely used in common REST protocols they don’t quite fit the R way of thinking about data.
RCX provides a collection of functions to integrate biological networks in CX format into analysis workflows. RCX adapts the aspect-oriented design in its data model, which consists of several aspects and sub-aspects, and corresponding properties, that are linked by internal IDs.

□ SingleCellMultiModal: Curated Single Cell Multimodal Landmark Datasets for R/Bioconductor
>> https://www.biorxiv.org/content/10.1101/2021.10.27.466079v1.full.pdf
SingleCellMultiModal, a suite of single-cell multimodal landmark datasets for benchmarking and testing multimodal analysis methods via the Bioconductor ExperimentHub package including CITE-Seq, ECCITE-Seq, SCoPE2, scNMT, 10X Multiome, seqFISH, and G&T.
For the integration of the 10x Multiome dataset, They used MOFA+ to obtain a latent embedding with contributom from both data modalities.
□ ddqc: Biology-inspired data-driven quality control for scientific discovery in single-cell transcriptomics
>> https://www.biorxiv.org/content/10.1101/2021.10.27.466176v1.full.pdf
data-driven QC (ddqc), an unsupervised adaptive quality control framework that performs flexible and data-driven quality control at the level of cell states while retaining critical biological insights and improved power for downstream analysis.
iterative QC, a revised paradigm to quality filtering best practices. It provides a data-driven quality control framework compatible with observed biological diversity.
□ IPJGL: Importance-Penalized Joint Graphical Lasso (IPJGL): differential network inference via GGMs
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab751/6414614
IPJGL, a novel importance-penalized joint graphical Lasso method for differential network inference based on the Gaussian graphical model with adaptive gene importance regularization.
DiNA focuses on gene interactions, which are more complex but can also reveal more information. a novel metric APC2 evaluates the interaction b/n a pair of genes for individual samples, which can be used in the downstream analyses of DiNA such as the gene-pair survival analysis.
□ CellexalVR: A virtual reality platform to visualize and analyze single-cell omics data
>> https://www.cell.com/iscience/fulltext/S2589-0042(21)01220-7
CellexalVR, an open-source virtual reality platform for the visualization and analysis of single-cell data. By placing all DR plots and associated metadata in VR is an immersive, feature-rich, and collaborative environment to explore and analyze scRNAseq experiments.
CellexalVR will also import cell surface marker intensities captured during index sorting/CITEseq and categorical metadata for cells and genes.

□ Filling gaps of genome scaffolds via probabilistic searching optical maps against assembly graph
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04448-2
This approach applies a sequential Bayesian updating to measure the similarity b/n optical maps and candidate contig paths. Using this similarity to guide path searching, It achieves higher accuracy than the existing “searching by evaluation” strategy that relies on heuristics.
nanoGapFiller aligns genome assembly contigs onto optical maps. The aligned contigs are further connected into scaffolds according to their order in the alignment.
nanoGapFiller uses a stochastic model to measure the similarity between a site sequence and any possible contig path, and then uses the probabilistic search technique to efficiently identify the contig path with the highest similarity.

□ Mix: A mixture model for signature discovery from sparse mutation data
>> https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00988-7
Mix algorithm for elucidating the mutational signature landscape of input samples from their (sparse) targeted sequencing data. Mix is a probabilistic model that simultaneously learns signatures and soft clusters patients, learning exposures per cluster instead of per sample.
Mix soft-clusters the patient’s mutations and takes a linear combination of all exposures according to their probability. With this, Mix also solves another problem of existing methods, where adding a new patient requires learning a new exposure vector for it.

□ NanoMethViz: An R/Bioconductor package for visualizing long-read methylation data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009524
NanoMethViz produces publication-quality plots to inspect the broad differences in methylation profiles of different samples, the aggregated methylation profiles of classes of genomic features, and the methylation profiles of individual long reads.
NanoMethViz converts results from methylation caller into a tabular format containing the sample name, 1-based single nucleotide chromosome position, log-likelihood-ratio of methylation and read name.

□ FASTAFS: file system virtualisation of random access compressed FASTA files
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04455-3
FASTAFS; a virtual layer between (random access) FASTA archives and read-only access to FASTA files and their guarenteed in-sync FAI, DICT and 2BIT files, through the File System in Userspace (FUSE).
FASTAFS guarantees in-sync virtualised metadata files and offers fast random-access decompression using bit encodings plus Zstandard (zstd).
FASTAFS, can track all its system-wide running instances, allows file integrity verification and can provide, instantly, scriptable access to sequence files and is easy to use and deploy.
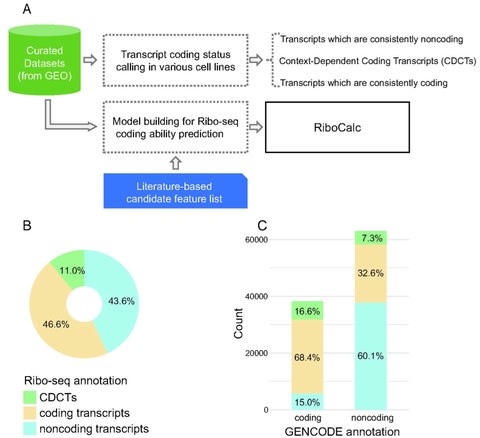
□ RiboCalc: Quantitative model suggests both intrinsic and contextual features contribute to the transcript coding ability determination in cells
>> https://www.biorxiv.org/content/10.1101/2021.10.30.466534v1.full.pdf
Ribosome Calculator (RiboCalc), an experiment-backed, data-oriented computational model for quantitatively predicting the coding ability (Ribo-seq expression level) of a particular human transcript. Features collected for RiboCalc model are biologically related to translation control.
RiboCalc not only makes quantitatively accurate predictions but also offers insight for sequence and transcription features contributing to transcript coding ability determination, shedding lights on bridging the gap between the transcriptome and proteome.
Large-scale analysis further revealed a number of transcripts w/ a variety of coding ability for distinct types of cells (i.e., context-dependent coding transcripts, CDCTs). A transcript’s coding ability should be modeled as a continuous spectrum with a context-dependent nature.
□ PopIns2: Population-scale detection of non-reference sequence variants using colored de Bruijn Graphs
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab749/6415820
PopIns2, a tool to discover and characterize Non-reference sequence (NRS) variants in many genomes, which scales to considerably larger numbers of genomes than its predecessor PopIns.
PopIns2 implements a scalable approach for generating a NRS variant call set. the paths through the graph have weights that may be used to compute a confidence score for each NRS. the traversal of the graph is trivially parallelizable on connected components of the graph.
□ Sub-Cluster Identification through Semi-Supervised Optimization of Rare-cell Silhouettes (SCISSORS) in Single-Cell Sequencing
>> https://www.biorxiv.org/content/10.1101/2021.10.29.466448v1.full.pdf
SCISSORS employs silhouette scoring for the estimation of heterogeneity of clusters and reveals rare cells in heterogenous clusters by implementing a multi-step, semi-supervised reclustering process.
SCISSORS calculates the silhouette score of each cell, which measures how well cells fit within their assigned clusters. The silhouette score estimates the relative cosine distance of each cell to cells in the same cluster versus cells in the closest neighboring cluster.
SCISSORS also enumerates several combinations of clustering parameters to achieve optimal performance by computing and comparing their silhouette coefficients.
□ Ideafix: a decision tree-based method for the refinement of variants in FFPE DNA sequencing data
>> https://academic.oup.com/nargab/article/3/4/lqab092/6412600
The Ideafix (deamination fixing) algorithm uses machine learning multivariate methods has the advantage over univariate methods that multiple descriptors can be tested simultaneously so that relationships between them can be exploited.
Assembled a collection of variant descriptors and evaluated the performance of five supervised learning algorithms for the classification of >1 600 000 variants, incl. both formalin-induced cytosine deamination artefacts and non-deamination variants, in order to arrive at Ideafix.
Unlike other methodologies that require multiple filtering steps and format conversion, the Ideafix algorithm is fully automatic.
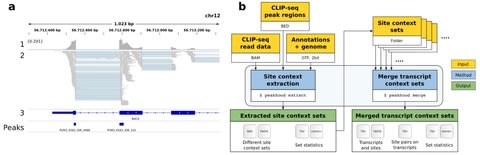
□ Peakhood: individual site context extraction for CLIP-seq peak regions
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab755/6420697
Peakhood, the first tool that utilizes CLIP-seq peak regions identified by peak callers, in tandem with CLIP-seq read information and genomic annotations, to determine which context applies, individually for each peak region.
Peakhood can merge single datasets into comprehensive transcript context site collections. The collections also include tabular data, for example to identify which sites on transcripts are in close distance, or if site distances decreased compared to the original genomic context.
□ decoupleR: Ensemble of computational methods to infer biological activities from omics data
>> https://www.biorxiv.org/content/10.1101/2021.11.04.467271v1.full.pdf
decoupleR, a Bioconductor package containing different statistical methods to extract these signatures within a unified framework. decoupleR allows the user to flexibly test any method with any resource.
decoupleR incorporates methods that take into account the sign and weight of network interactions. With a common syntax. for types of omics datasets, and knowledge sources, it facilitates the exploration of different approaches and can be integrated in many workflows.
□ Re-expressing coefficients from regression models for inclusion in a meta-analysis
>> https://www.biorxiv.org/content/10.1101/2021.11.02.466931v1.full.pdf
When the distribution of exposure is skewed, the re-expression methods examined are likely to give biased results. The bias varied by method, the direction of the re-expression, skewness, influential observations, and in some cases, the median exposure.
Meta-analysts using any of these re-expression methods may want to consider the uncertainty, the likely direction and degree of bias, and conduct sensitivity analyses on the re-expressed results.
□ Bringing Light Into the Dark: A Large-scale Evaluation of Knowledge Graph Embedding Models under a Unified Framework
>> https://ieeexplore.ieee.org/document/9601281
large-scale benchmarking on four datasets with several thousands of experiments and 24,804 GPU hours of computation time.
the combination of model architecture, training approach, loss function, and the explicit modeling of inverse relations is crucial for a model's performance and is not only determined by its architecture.

□ EMBL
>> https://www.embl.org/topics/cop26/
EMBL is proud to have been formally admitted as an official Observer organisation by the 26th session of the UN Conference of the Parties @COP26.
We look forward to contributing further to the process of the UN's Framework Convention on Climate Change.
□ Rob Fin
>> https://twitter.com/robdfinn/status/1456936786547052546?s=21
@Google currently talking about the importance of data, ML and high throughput computing solutions to understand deforestation #COP26 image data, geo spatial data, monitoring, all sounds familiar to what we do at @emblebi

□ Making Common Fund data more Findable: Catalyzing a Data Ecosystem
>> https://www.biorxiv.org/content/10.1101/2021.11.05.467504v1.full.pdf
The CFDEs federation system is centered on a metadata catalog that ingests metadata from individual Common Fund Program Data Coordination Centers into a uniform metadata model that can then be indexed and searched from a centralized portal.
This uniform Crosscut Metadata Model (C2M2), supports the wide variety of data set types and metadata terms used by the individual and is designed to enable easy expansion to accommodate new datatypes.

□ hybpiper-rbgv and yang-and-smith-rbgv: Containerization and additional options for assembly and paralog detection in target enrichment data
>> https://www.biorxiv.org/content/10.1101/2021.11.08.467817v1.full.pdf
HybPiper-RBGV: containerised and pipelined using Singularity and Nextflow. hybpiper-rbgv creates two output folders, one with all supercontigs and one with suspected chimeras (using read-mapping to supercontigs and identification of discordant read-pairs) removed.
The Maximum Inclusion algorithm iteratively extracts the largest subtrees from an unrooted gene tree. The Monophyletic Outgroups algorithm removes all genes in which is non-monophyletic. These alignments are ready for phylogenetic analysis either separately or after concatenation.
□ Emulating complex simulations by machine learning methods
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04354-7
a multi-output dynamic simulation model, which, given a set of input, it generates the dynamics of a multivariate vector over a given time horizon.
A pitfall of this method is that it does not exploit the dynamics of the simulated process while relying just on the initial condition. This approach does not fit well those cases in which the modelled process has a large variability as for instance in stochastic simulations.

□ Low-input ATAC&mRNA-seq protocol for simultaneous profiling of chromatin accessibility and gene expression
>> https://star-protocols.cell.com/protocols/968
a simple, fast, and robust protocol (low-input ATAC&mRNA-seq) to simultaneously generate ATAC-seq and mRNA-seq libraries from the same cells in limited cell numbers by coupling a simplified ATAC procedure using whole cells with a novel mRNA-seq approach.
that features a seamless on-bead process including direct mRNA isolation from the cell lysate, solid-phase cDNA synthesis, and direct tagmentation of mRNA/cDNA hybrids for library preparation.










※コメント投稿者のブログIDはブログ作成者のみに通知されます