









無慈悲な重力に燃立つカオスの縁で、我々は意思に依って立ち、智慧を道標としている。回る星の下で互いに時計を合わせ、運命を書き換えるその瞬間まで、流れ落ちる砂から這い上がろうと足掻き続けている。心とは光の動態そのものであるからだ。

□ Phantom Purge: Statistical modeling, estimation, and remediation of sample index hopping in multiplexed droplet-based single-cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/24/617225.full.pdf
a probabilistic model that formalizes the phenomenon of index hopping and allows the accurate estimation of its rate.
Application of the proposed model to several multiplexed datasets suggests that the sample index hopping probability for a given read is approximately 0.008, an arguable low number, even though, counter-intuitively, it can give rise to a large fraction of phantom molecules.


□ psupertime: supervised pseudotime inference for single cell RNA-seq data with sequential labels
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/29/622001.full.pdf
psupertime, a supervised pseudotime approach which outperforms benchmark pseudotime methods by explicitly using the sequential labels as input.
A non-zero ordering coefficient indicates that a gene was relevant to the label sequence. psupertime attains a test accuracy of 83% over the 8 possible labels, using 82 of the 827 highly variable genes.
a pseudotime value for each individual cell, obtained by multiplying the log gene expression values by the vector of coefficients; and a set of values along the pseudotime axis indicating the thresholds between successive sequential labels.


□ CANDID: Time-resolved genome-scale profiling reveals a causal expression network
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/01/619577.full.pdf
using hard-thresholding to remove (i.e., set equal to zero) the majority of values in the dataset, leaving ~100,000 timecourses with coherent, biologically-feasible patterns of variability.
CANDID (Causal Attribution Networks Driven by Induction Dynamics) for revealing genome-wide causal relationships without incorporating prior information, resulted in the prediction of multiple transcriptional regulators that were validated experimentally.
By aggregating all timecourses, we can more confidently identify which regulator(s) are acting in each individual timecourse by finding the parsimonious set of regulators whose abundances account for each gene’s expression variability.

□ Explosive synchronization in frequency displaced multiplex networks
>> https://aip.scitation.org/doi/full/10.1063/1.5092226
a close relationship between structure and dynamics in the process of synchronization in complex networks has been the object of study for a long time;
however, it has proved to be particularly important in the case of the “explosive synchronization,” where the ensemble reaches suddenly to a fully coherent state through a discontinuous, irreversible first-order like transition, often in the presence of a hysteresis loop.

□ NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz240/5424072
generic PPI networks, users can now create cell-type or tissue specific PPI networks, gene regulatory networks, gene co-expression networks as well as networks for toxicogenomics and pharmacogenomics.
The resulting networks can be customized and explored in VR space. a global enrichment network in which nodes represent functions and edges are determined by the overlap ratio between genes associated with the two functions. These nodes are implemented as meta-nodes.
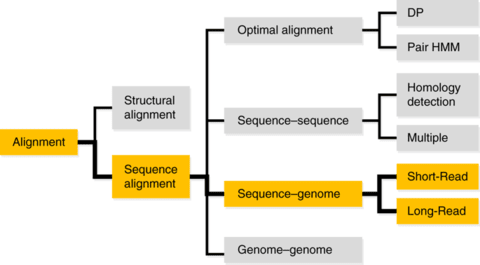
□ Accurate high throughput alignment via line sweep-based seed processing
>> https://www.nature.com/articles/s41467-019-09977-2
An algorithmic scheme, two line sweep-based techniques called “strip of consideration” and “seed harmonization”. It performs alignments by completing the following three stages: Seeding, seed processing and dynamic programming.
The FMD-index allows the computation of supermaximal exact matches (SMEMs). this approach does not rely on specially tailored data structures and it can be described concisely in pseudocode.
The overall time complexity of the SoC computational is limited by the complexity of an initial seed sorting. If the index used for seed generation is able to deliver the seeds in correct order, then the SoC can be computed in a single pass in linear time.
□ Steane-Enlargement of Quantum Codes from the Hermitian Curve
>> https://arxiv.org/pdf/1904.10007v1.pdf
A k-dimensional quantum code of length n over Fq is a qk-dimensional subspace of the Hilbert space Cqn. This space is subject to phase-shift errors, bit-flip errors, and combinations thereof.
For codes of sufficiently large dimension, however, the Goppa bound does not give the true minimal distance, and the order bound for dual codes and for primary codes give more information on the minimal distance of the codes.
the construction of quantum codes by applying Steane- enlargement to codes from the Hermitian curve. By using the Steane-enlargement technique, the minimal distance dx can be increased by one, yielding a symmetric quantum code of the same dimension.

□ Coxeter submodular functions and deformations of Coxeter permutahedra https://arxiv.org/pdf/1904.11029.pdf
There are natural Coxeter analogs of compositions, graphs, matroids, posets, and clusters, and can observe that they are all part of this framework of deformations of Φ-permutahedra. generalized Φ-permutahedra should be an important example of a new kind of algebraic structure: a Coxeter Hopf monoid.
□ A weighted sequence alignment strategy for gene structure annotation lift over from reference genome to a newly sequenced individual
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/22/615476.full.pdf
the natural variation alleles expression level of apoptosis death and defence response related genes might could be better quantified using GEAN.
GEAN could be used to refine the functional annotation of genetic variants, annotate de novo assembly genome sequence, detect syntenic blocks, improve the quantification of gene expression levels using RNA-seq & genomic variants encoding for population genetic analysis.
a zebraic dynamic programming (ZDP) by providing different weights to different genetic features to refine the gene structure lift over. ZDP is a semi-global sequence alignment algorithm & software to infer about the gene structure of non-reference accession/line w/ an algorithm.

□ Syntenizer 3000: Synteny-based analysis of orthologous gene groups
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/25/618678.full.pdf
a novel algorithm for measuring the degree of synteny shared between two genes and successfully disambiguate gene groups.
The large discrepancy between synteny scores of the paralog selected as the true syntelog, and the other candidates is a strong indicator of the viability of our synteny based disambiguation method for the dataset.

□ Metascape provides a biologist-oriented resource for the analysis of systems-level datasets:
>> https://www.nature.com/articles/s41467-019-09234-6
Metascape combines functional enrichment, interactome analysis, gene annotation, and membership search to leverage over 40 independent knowledgebases within one integrated portal.
Metascape utilizes the well-adopted hypergeometric test and Benjamini-Hochberg p-value correction algorithm to identify all ontology terms that contain a statistically greater number of genes.
□ Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads
>> https://www.nature.com/articles/s41592-019-0394-y
D-NAscent is aa sequencing method for the measurement of replication fork movement on single molecules by detecting nucleotide analog signal currents on extremely long nanopore traces.
D-NAscent detects the differences in BrdU incorporation frequency across individual molecules to reveal the location of active replication origins, fork direction, termination sites, and fork pausing/stalling events.
□ Direct Comparative Analysis of 10X Genomics Chromium and Smart-seq2
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/22/615013.full.pdf
The composite of Smart-seq2 data also resembled bulk RNA-seq data better. For 10X-based data, we observed higher noise for mRNA in the low expression level. Despite the poly(A) enrichment, approximately 10-30% of all detected transcripts by both platforms were from non-coding genes, with lncRNA accounting for a higher proportion in 10X.
□ TransLiG: a de novo transcriptome assembler that uses line graph iteration
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1690-7
TransLiG, a new de novo transcriptome assembler, which is able to integrate the sequence depth and pair-end information into the assembling procedure by phasing paths and iteratively constructing line graphs starting from splicing graphs.
TransLiG accurately links the in-coming and out-going edges at each node via iteratively solving a series of quadratic programmings, which are optimizing the utilizations of the paired-end and sequencing depth information.

□ SLant: Predicting synthetic lethal interactions using conserved patterns in protein interaction networks
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006888
SLant (Synthetic Lethal analysis via Network topology), a computational systems approach to predicting human synthetic lethal interactions that works by identifying and exploiting conserved patterns in protein interaction network topology both within and across species. These features comprise both node-wise distance and pairwise topological PPI parameters & gene ontology, and identifies a large cohort of candidate human synthetic lethal pairs which are available with the consensus predictions for all the model organisms in the Slorth database.

□ BAGEA: A Framework for Integrating Directed and Undirected Annotations to Build Explanatory Models of cis-eQTL Data
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/26/619452.full.pdf
Bayesian Annotation Guided eQTL Analysis (BAGEA), a variational Bayes framework to model cis-eQTLs using directed and undirected genomic annotations.
BAGEA can directly model phenomena relevant to genetic architecture, such as the relatively larger impact of SNPs close to the TSS on directed annotations compared to that of distal SNPS, making BAGEA mores useful for predictive modeling.
□ Unsupervised machine learning in atomistic simulations, between predictions and understanding
>> https://aip.scitation.org/doi/full/10.1063/1.5091842
Statistical learning will contribute to the increase in complexity by making it possible to side-step time-consuming electronic structure calculations and obtain accurate interatomic potentials that can be evaluated on large systems and for long trajectories.
example of the synergy between supervised and unsupervised learning tasks involves the use of regression techniques to reconstruct (high-dimensional) free-energy surfaces, which mitigates the problem of the curse of dimensionality when performing essentially a density estimation.
□ Insights into protein sequencing with an α-Hemolysin nanopore by atomistic simulations
>> https://www.nature.com/articles/s41598-019-42867-7
an extensive set of non-equilibrium all-atom MD simulations (≃8μs in total) to calculate the current levels associated to four different neutral homopeptides. an equilibrium quantity derived from continuum quasi-1D argument and indicated as “pore clogging estimator” is linearly correlated to the measured current blockages from non-equilibrium runs.

□ Calibrating seed-based heuristics to map short DNA reads
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/25/619155.full.pdf
a theory to estimate the probability that reads are mapped to a wrong location due to limitations at the seeding step. the properties of simple exact seeds, skip-seeds and MEM seeds (Maximal Exact Match).
The main innovation of this work is to use concepts from analytic combinatorics to represent reads as abstract sequences, and to specify their generative function to estimate the probabilities of interest.
□ RNA-align: quick and accurate alignment of RNA 3D structures based on size-independent TM-scoreRNA
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz282/5480133
RNA-align seeks optimal nucleotide-to-nucleotide alignments based on a heuristic dynamic programming iteration process, assisted by distance-based secondary structure assignments.
The major advantage of RNA-align lies at the quick convergence of the heuristic alignment iterations and the coarse-grained secondary structure assignment, both of which are crucial to the speed and accuracy of RNA structure alignments.
□ Another Look at Matrix Correlations
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz281/5480130
a principled approach based on the matrix decomposition generates three trace-independent parts for every matrix.
the decomposition results in the removal of high correlation bias and the dependence on the sample number intrinsic to the RV coefficient.
□ PHANOTATE: A novel approach to gene identification in phage genomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz265/5480131
PHANOTATE, a novel method for gene calling specifically designed for phage genomes. While the compact nature of genes in phages is a problem for current gene annotators, exploit this property by treating a phage genome as a network of paths: where open reading frames are favorable, and overlaps and gaps are less favorable, but still possible, and represent this network of connections as a weighted graph, and use dynamic programming to find the optimal path.

□ SuperVec: Learning supervised embeddings for large scale sequence comparisons
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/26/620153.full.pdf
SuperVec provides flexibility to utilize meta-information along with the contextual information present in the sequences to generate their embeddings. The SuperVec approach is extended further through H-SuperVec, a tree-based hierarchical method which learns embeddings across a range of feature spaces based on the class labels and their exclusive and exhaustive subsets.
□ RTDT: A new method for inferring timetrees from temporally sampled molecular sequences
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/26/620187.full.pdf
two non-Bayesian methods (RTDT and Least Squares Dating [LSD]) to perform similar to or better than the Bayesian approaches available in BEAST and MCMCTree programs.
RTDT estimates pathogen timetrees based on the relative rate framework underlying the RelTime approach. RTDT performed better than the other methods for the estimation of divergence times at deep node in phylogenies where evolutionary rates were autocorrelated.
□ CESAR: Coding Exon-Structure Aware Realigner: Utilizing Genome Alignments for Comparative Gene Annotation
>> https://link.springer.com/protocol/10.1007%2F978-1-4939-9173-0_10
CESAR 2.0 is a method to realign coding exons or genes to DNA sequences using a Hidden Markov Model.
CESAR 2.0 provides a new gene mode that re-aligns entire genes at once. CESAR 2.0 is 77X times faster on average (132X times faster for large exons) and requires 30-times less memory.
□ ORCA: Genomics Research Container Architecture
>> https://github.com/bcgsc/orca
ORCA provides a comprehensive bioinformatics container environment, which may be installed with a single command, and includes hundreds of pre-compiled and configured bioinformatics tools.
□ GenMap: Fast and Exact Computation of Genome Mappability
>> https://www.biorxiv.org/content/biorxiv/early/2019/04/26/611160.full.pdf
GenMap computes the mappability of genomes up to e errors, which is based on the C++ sequence analysis library SeqAn. GenMap is a fast and exact algorithm to compute the (k,e)-mappability. Its inverse, the (k,e)- frequency counts the number of occurrences of each k-mer with up to e errors in a sequence.
□ Expression estimation and eQTL mapping for HLA genes with a personalized pipeline
>> https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1008091
the HLA-personalized pipeline is more accurate than conventional mapping, and apply the tool to reanalyze RNA-seq data from the GEUVADIS Consortium.
□ scMatch: a single-cell gene expression profile annotation tool using reference datasets
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz292/5480299
scMatch directly annotates single cells by identifying their closest match in large reference datasets. using this strategy to annotate various single-cell datasets and evaluated the impacts of sequencing depth, similarity metric and reference datasets.

□ SPARK-MSNA: Efficient algorithm on Apache Spark for aligning multiple similar DNA/RNA sequences with supervised learning
>> https://www.nature.com/articles/s41598-019-42966-5
SPARK-MSNA algorithm provides linear complexity O(m) compared to O(m2). The overall complexity of SPARK-MSNA is 𝑂(𝑚)+𝑂(𝑛2𝑚)+𝑂(𝑛)+𝑂(𝑘)+𝑂(𝑘)+𝑂(𝑛𝑚). Comparing with state-of-the-art algorithms (e.g., HAlign II).
SPARK-MSNA provided 50% improvement in memory utilization in processing human mitochondrial genome (mt. genomes, 100x, 1.1. GB) with a better alignment accuracy in terms of average SP score and comparable execution time. Key characteristics of the proposed algorithm include, Suffix tree data structure for storing input sequences and identifying common substrings between sequences, A knowledge base and nearest neighbor learning layer to guide the pairwise alignment,
Modified dynamic programming algorithm to perform pairwise alignments at each stage in order to reduce the memory and execution time of alignments and Parallelization using MapReduce method for suffix tree construction and pairwise alignment to further improve the execution time.

□ Trans Effects on Gene Expression Can Drive Omnigenic Inheritance
>> https://www.cell.com/cell/fulltext/S0092-8674(19)30400-3
a formal model in which genetic contributions to complex traits are partitioned into direct effects from core genes and indirect effects from peripheral genes acting in trans. This model proposes a framework for understanding key features of the architecture of complex traits.
The most heritability is driven by weak trans-eQTL SNPs, whose effects are mediated through peripheral genes to impact the expression of core genes.
□ Trajectory-based differential expression analysis for single-cell sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/02/623397.full.pdf
a powerful generalized additive model framework based on the negative binomial distribution that allows flexible inference of within-lineage differential expression by detecting associations between gene expression and pseudotime over an entire lineage.
by comparing gene expression between points/regions within the lineage and between-lineage differential expression by comparing gene expression between lineages over the entire lineages or at specific points/regions. By incorporating observation-level weights, the model additionally allows to account for zero inflation, commonly observed in single-cell RNA-seq data from full-length protocols.
□ Genes with High Network Connectivity Are Enriched for Disease Heritability
>> https://www.cell.com/ajhg/fulltext/S0002-9297(19)30116-8
For each gene network, these pathway+network annotations were strongly significantly enriched for the corresponding traits. the enrichments were largely explained by the baseline-LD model.
gene network connectivity is highly informative for disease architectures, but the information in gene networks may be subsumed by regulatory annotations, emphasizing the importance of accounting for known annotations.

□ DNA energy constraints shape biological evolutionary trajectories
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/03/625681.full.pdf
the biological information contained within a dsDNA molecule, in terms of a linear sequence of nucleotides, has been considered the main target of the evolution, in this information-centred perspective, certain DNA sequence symmetries are difficult to explain. these patterns can emerge from the physical peculiarities of the dsDNA molecule itself and the maximum entropy principle alone.
the physical properties of the dsDNA are the hard drivers of the overall DNA sequence architecture, whereas the biological selective processes act as soft drivers, which only under extraordinary circumstances overtake the overall entropy content of the genome.

□ Multiplexed dissection of a model human transcription factor binding site architecture
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/02/625434.full.pdf
the number and affinity of c-AMP Response Elements (CREs) within regulatory elements largely determines overall expression, and this relationship is shaped by the proximity of each CRE to the downstream promoter.
compare library expression between an episomal MPRA and a new, genomically-integrated MPRA in which a single synthetic regulatory element is present per cell at a defined locus. these largely recapitulate each other although weaker, non-canonical CREs exhibited greater activity in the genomic context.
□ Using Deep Learning to Annotate the Protein Universe
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/03/626507.full.pdf
a deep learning model that learns the relationship between unaligned amino acid sequences and their functional classification across all 17929 families of the Pfam database.
Using the Pfam seed sequences, and establish a rigorous benchmark assessment and find a dilated convolutional model that reduces the error of both BLASTp and pHMMs by a factor of nine.
□ larsjuhljensen:
since genes/proteins are evolutionarily related, random partitioning does not give you an independent test set.
□ SavvyCNV: genome-wide CNV calling from off-target reads
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/03/617605.full.pdf
Using truth sets generated from genome sequencing data and MLPA, SavvyCNV outperformed four state-of-the-art CNV callers at calling CNVs using off-target reads. We then identified clinically relevant CNVs from a targeted panel using SavvyCNV.
□ Identification of genes under dynamic post-transcriptional regulation from time-series epigenomic data
>> https://www.futuremedicine.com/doi/10.2217/epi-2018-0084
time-series profiles of chromatin immunoprecipitation-seq data of histone modifications from differentiation of mesenchymal progenitor cells to predict gene expression levels at five time points in both lineages and estimated the deviation of those predictions from the RNA-seq measured expression levels using linear regression.
Clustering mRNAs according to their stability dynamics allows identification of post-transcriptionally coregulated mRNAs and their shared regulators through sequence enrichment analysis.
□ SysGenetiX: A model to decipher the complexity of gene regulation
>> https://www.sciencedaily.com/releases/2019/05/190502143513.htm
SysGenetiX (UNIGE/UNIL) aimed to investigate the regulatory elements, as well as the manifold interactions between them and with genes, with the ultimate goal of understanding the mechanisms that render some people more predisposed to manifesting particular diseases than others. By incorporating the complexity of the genome into a single model, SysGenetiX provides a tree of correlations of all regulatory elements across the whole genome.
Every node of this tree can then be analysed to summarize the effects of that node as well as the variability of all regulatory elements below that could be relevant to a certain phenotype.










※コメント投稿者のブログIDはブログ作成者のみに通知されます