









我々は星屑から産まれ、星屑を集め、星屑へと還る。余燼を焚べた炉であり、煤けた情報の断片であり、己が綴られたコンテクストを読み取る術はない。しかし視えるのだ。擦れ合う骨が血と肉を運ぶように、天蓋の向こうで沈黙する岩と焦げたガスとの狭間に、我々を繋ぎ止めている一本の鎖が
We are made of stardust gather stardust, and return to stardust. We are a furnace that stokes the remaining embers, fragments of sooty information, with no means to decipher the context we've woven for ourselves.
Yet, we can see. Just as rubbing bones transport blood and flesh, amidst the silence of rocks and scorched gases beyond the canopy, there exists a single chain that binds us.

□ CodonBERT: Large Language Models for mRNA Design and Optimization
>> https://www.biorxiv.org/content/10.1101/2023.09.09.556981v1
CodonBERT, an LLM which extends the BERT model and applies it to the language of mRNAs. CodonBERT uses a multi-head attention transformer architecture framework. The pre-trained model can also be generalized to a diverse set of supervised learning tasks.
CodonBERT is pre-trained using 10 million mRNA coding sequences spanning an evolutionarily diverse set of organisms. CodonBERT takes the coding region as input using codons as tokens, and outputs an embedding that provides contextual codon representations.

□ scAce: an adaptive embedding and clustering method for single-cell gene expression data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad546/7261512
scAce constructs a VAE network to learn smoother low-dimensional embeddings compared with those methods based on traditional autoencoders. It utilizes a data-adaptive clustering approach based on the idea of cluster merging.
scAce iteratively performs network update and cluster merging based on the initial VAE network. scAce decides if a pair of clusters should be merged into a single cluster by comparing inter-cluster and intra-cluster distances.

□ scEval: Evaluating the Utilities of Large Language Models in Single-cell Data Analysis
>> https://www.biorxiv.org/content/10.1101/2023.09.08.555192v1
scEval (Single-cell Large Language Model Evaluation), a systematic evaluation of the effects of hyper-parameters, initial settings, and stability for training single-cell LLMs. Evaluating the performance of single-cell LLMs - scGPT, Geneformer, scBERT, CellLM and tGPT.
scGPT is capable of performing zero-shot learning tasks. For the Cell Lines dataset, the zero-shot learning approach even achieved the highest score, indicating that it can be an effective method for certain datasets.
GEARS was gen-erall better than scGPT. For the data simulation task, scGPT did not perform very well, which suggests that LLMs are remembering things rather than making inferences or generating enough novel information.

□ Autoturbo-DNA: Turbo-Autoencoders for the DNA data storage channel
>> https://www.biorxiv.org/content/10.1101/2023.09.15.557887v1
Autoturbo-DNA, an end-to-end autoencoder framework that combines the TurboAE principles with an additional pre-processing decoder, DNA data storage channel simulation, and constraint adherence check.
Autoturbo-DNA supports various Neural-Network architectures. Autoturbo-DNA trains encoder-transcoder-decoder models for DNA data storage. Autoturbo-DNA reconstructs performance close to single sequence non-NN error correction and constrained codes for DNA data storage.
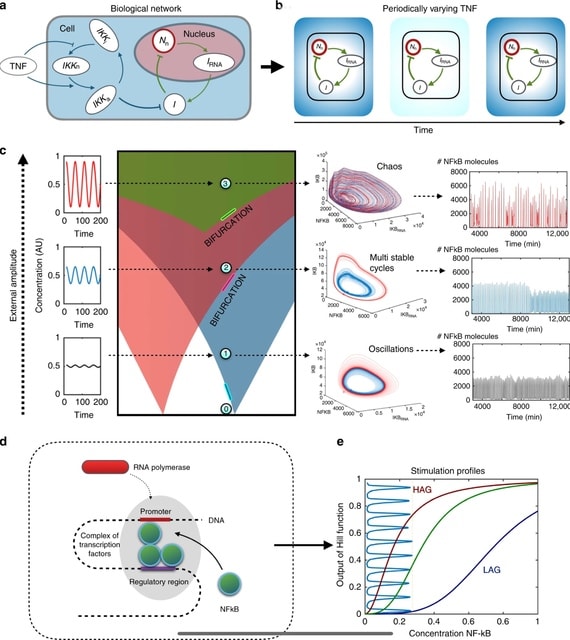
□ On chaotic dynamics in transcription factors and the associated effects in differential gene regulation
>> https://www.nature.com/articles/s41467-018-07932-1
All deterministic simulations were performed by numerically integrating the dynamical equations using the Runge–Kutta fourth-order method, and for optimisation reasons, some of the equations were simulated using Euler integration.
Chaotic dynamics has far been underestimated as a means for controlling genes. They tested for chaos by calculating the divergence of trajectories that started at almost identical initial points. NF-κB driven by sufficiently large TNF amplitudes will exhibit deterministic chaos.

□ ZINBMM: a general mixture model for simultaneous clustering and gene selection using single-cell transcriptomic data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03046-0
ZINBMM, a zero-inflated negative binomial mixture model for scRNA-seq data clustering that can comprehensively account for the unique problems of batch effects, dropout events, and high dimensionality. ZINBMM directly applies to the raw counts without any transformation.
The mixture model with biological effects of genes being modelled using cell type-specific mean parameters is developed to accommodate heterogeneity, which achieves soft clustering and has the advantage of more meaningful probabilistic interpretations.
ZINBMM can accommodate zero-expressed gene counts and correct the confounding batch effects by introducing corresponding parameterisation. ZINBMM performs feature selection by imposing penalisation on the differences between cluster-specific and global mean values.

□ Borzoi: Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation
>> https://www.biorxiv.org/content/10.1101/2023.08.30.555582v1
Borzoi learns to predict cell- and tissue-specific RNA-seq coverage from DNA sequence. Borzoi isolates and accurately scores variant effects across multiple layers of regulation, including transcription, splicing, and polyadenylation.
Borzoi uses the core Enformer architecture, which includes a tower of convolution- and subsampling blocks followed by a series of self-attention blocks operating at 128 bp resolution embedding vectors.

□ scover: Predicting the impact of sequence motifs on gene regulation using single-cell data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03021-9
scover infers regulatory motifs that are predictive of the signal associated with a set of sequences using a neural network consisting of a single convolutional layer, an exponential linear unit, global max pooling, and a linear layer with bias term.
Scover takes as input a set of one-hot encoded sequences, e.g., promoters or distal enhancers, along with measurements of their activity, e.g., expression levels of the associated genes or accessibility levels of the enhancers.

□ GENIX: Comparative Analysis of Association Networks Using Single-Cell RNA Sequencing Data Reveals Perturbation-Relevant Gene Signatures
>> https://www.biorxiv.org/content/10.1101/2023.09.11.556872v1
GENIX (Gene Expression Network Importance eXamination), a novel platform for constructing gene association networks, equipped with an innovative network-based comparative model to uncover condition-relevant genes.
By leveraging this probabilistic graphical model, GENIX faithfully differentiates between direct and indirect connections while remaining immune to neglecting novel interactions, a common downside of reference-guided network construction methods.
GENIX uses a systematic module identification and analysis approach, and a two-dimensional quantitative metric, providing a more comprehensive understanding of changes in gene essentiality within the network upon perturbation.

□ NetAn: A Python Toolbox Leveraging Network Topology for Comprehensive Gene Annotation Enrichments
>> https://www.biorxiv.org/content/10.1101/2023.09.05.556339v1
NetAn (the Network Annotation Enrichment package), which takes a list of genes and uses network-based approaches such as network clustering and inference of closely related genes to include local neighbours.
NetAn draws the adjacency matrix of the input gene set from the loaded network, and applies either K-means clustering, maximal clique identification, or the extraction of separated network components to sort genes into individual sets.
NetAn has a functionality where the average shortest path length between all gene cluster pairs is computed and compared to the average path length of the loaded network. NetAn randomly samples pairs in batches until the mean converges.

□ PAN-GWES: Pangenome-spanning epistasis and co-selection analysis via de Bruijn graphs
>> https://www.biorxiv.org/content/10.1101/2023.09.07.556769v1
PAN-GWES, a phenotype- and alignment-free method for discovering co-selected and epistatically interacting genomic variation from genome assemblies covering both core and accessory parts of genomes.
PAN-GWES uses a compact coloured de Bruijn graph to approximate the intra-genome distances between pairs of loci. PAN-GWES leverages the computational efficiencies of the SpydrPick algorithm to rapidly calculate the pairwise MI values of millions of unitigs pairs.
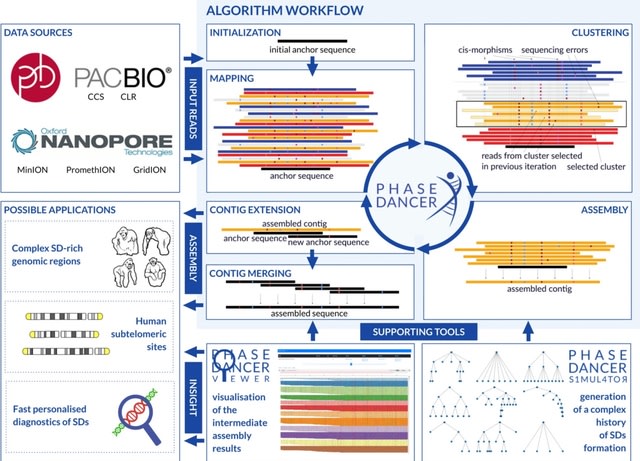
□ PhaseDancer: a novel targeted assembler of segmental duplications unravels the complexity of the human chromosome 2 fusion going from 48 to 46 chromosomes in hominin evolution
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03022-8
PhaseDancer, a novel, fast, and robust assembler that follows a locally-targeted approach to resolve SD-rich complex genomic regions. The tool is designed to work with long-reads (ONT, PacBio) and tuned for error-prone data.
PhaseDancer enables the extension of a user-provided initial sequence contig even from complex genomic regions. PhaseDancer generates contigs with the fragments repeated up to several dozens times in the genome with at least 0.1% divergence.
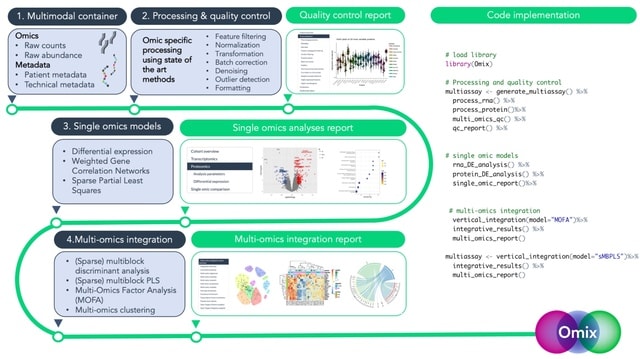
□ Omix: A Multi-Omics Integration Pipeline
>> https://www.biorxiv.org/content/10.1101/2023.08.30.555486v1
Omix is built on four consecutive blocks, (1) preparation of the multimodal container, (2) processing and quality control, (3) single omic analyses, and (4) multi-omics vertical integration,
The modular framework of Omix enables the storage of analysis parameters and results from different algorithms within the same object, facilitating easy comparison of outputs. This design also allows for the incorporation of additional integrative models as the field progresses.

□ CellsFromSpace: A versatile tool for spatial transcriptomic data analysis with reference-free deconvolution and guided cell type/activity annotation
>>
CellsFromSpace decomposes spatial transcriptomic data into components that represent distinct cell types or activities. The direct annotation of components, allows users to identify and isolate cell populations in the latent space, even when they overlap.
CellsFromSpace overcomes some of the limitation of Latent Dirichlet Allocation. CFS is based on the independent component analysis (ICA), a blind source separation technique that attempts to extract sources from a mixture of these sources.

□ Scoring alignments by embedding vector similarity
>> https://www.biorxiv.org/content/10.1101/2023.08.30.555602v1
The E-score project focuses on computing Global-regular and Global-end-gap-free alignment between any two protein sequences using their embedding vectors computed by stat-of-art pre-trained models.
Instead of a fixed score between two pairs of amino acids, they use the cosine similarity between the embedding vectors of two amino acids and use it as the context-dependent score.

□ AliSim-HPC: parallel sequence simulator for phylogenetics
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad540/7258693
AliSim-HPC is highly efficient and scalable, which reduces the runtime to simulate 100 large gap-free alignments (30,000 sequences of one million sites) from over one day to 11 minutes using 256 CPU cores from a cluster with 6 computing nodes, a 153-fold speedup.
AliSim-HPC parallelizes the simulation process at both multi-core and multi-CPU levels using the OpenMP and MPI libraries. AliSim-HPC employs The Scalable Parallel Random Number Generators Library (SPRNG) and requires users to specify a random number generator seed.

□ MuLan-Methyl—multiple transformer-based language models for accurate DNA methylation prediction
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad054/7230465
MuLan-Methyl, a deep learning framework for predicting DNA methylation sites, which is based on 5 popular transformer-based language models. The framework identifies methylation sites for 3 different types of DNA methylation: N6-adenine, N4-cytosine, and 5-hydroxymethylcytosine.
Each of the employed language models is adapted to the task using the “pretrain and fine-tune” paradigm. Pretraining is performed on a custom corpus of DNA fragments and taxonomy lineages. Fine-tuning aims at predicting the DNA methylation status of each type.

□ SpatialDDLS: An R package to deconvolute spatial transcriptomics data using neural networks
>> https://www.biorxiv.org/content/10.1101/2023.08.31.555677v1
SpaDalDDLS leverages single-cell RNA sequencing data to simulate mixed transcripDonal profiles with predefined cellular composiDon, which are subsequently used to train a fully-connected neural network to uncover cell type diversity within each spot.
SpatialDDLS offers the option to keep only those genes present in a specified number of slides. These steps aim to expedite subsequent steps by avoiding the consideration of the entire noisy expression matrix.

□ spaTrack: Inferring cell trajectories of spatial transcriptomics via optimal transport analysis
>> https://www.biorxiv.org/content/10.1101/2023.09.04.556175v1
spaTrack, a trajectory inference method incorporating both expression and distance cost of cell transition. spaTrack utilizes Optimal Transport (OT) as a foundation to infer the transition probability between cells of ST data in a single sample.
spaTrack models the fate of a cell as a function of expression profile along temporal intervals driven by TF. spaTrack can construct a dynamic map of cell migration and differentiation across all tissue sections, providing a comprehensive view of transition behavior over time.

□ SCGP: Characterizing tissue structures from spatial omics with spatial cellular graph partition
>> https://www.biorxiv.org/content/10.1101/2023.09.05.556133v1
Spatial Cellular Graph Partitioning (SCGP) is a fast and flexible method designed to identify the anatomical and functional units in human tissues. It can be effectively applied to both spatial proteomics and transcriptomics measurements.
SCGP-Extension, which enables the generalization usage of extending a set of reference tissue structures to previously unseen query samples. SCGP-Extension can address challenges ranging from experimental artifacts, batch effects, to disease condition differences.
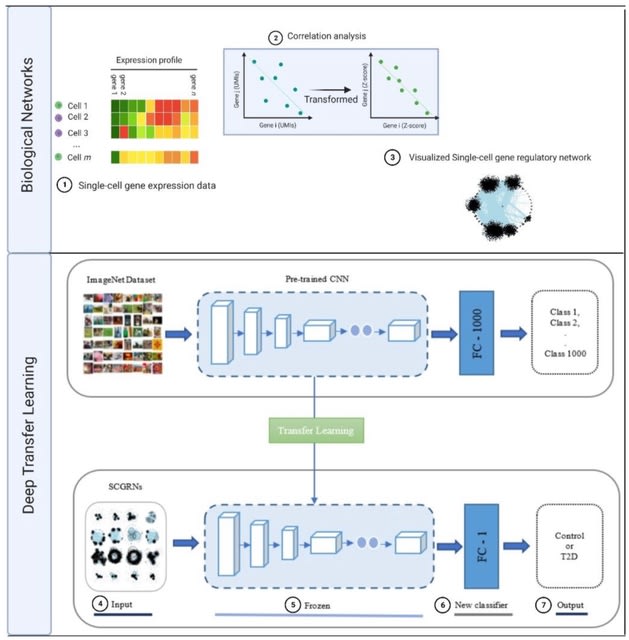
□ A novel interpretable deep transfer learning combining diverse learnable parameters for improved prediction of single-cell gene regulatory networks
>> https://www.biorxiv.org/content/10.1101/2023.09.07.556481v1
In terms of the TFt-based models, they keep weights of the bottom layers in the feature extraction part of pre-trained models unchanged while modifying weights in the proceeding layers including the densely connected classifier according to the Adam optimizer.
The densely connected classifier was altered to deal w/ the binary class classification problem pertaining to distinguishing between healthy controls and T2D SCGRN images. It can be seen that updating model weight parameters is done through the training w/ the Adam optimizer.

□ CS-CORE: Cell-type-specific co-expression inference from single cell RNA-sequencing data
>> https://www.nature.com/articles/s41467-023-40503-7
CORE (cell-type-specific co-expressions) models the unobserved true gene expression levels as latent variables, linked to the observed UMI counts through a measurement model that accounts for both sequencing depth variations and measurement errors.
CS-CORE implements a fast and efficient iteratively re-weighted least squares approach for estimating the true correlations between underlying expression levels, together with a theoretically justified statistical test to assess whether two genes are independent.

□ μ-PBWT: a lightweight r-indexing of the PBWT for storing and querying UK Biobank Data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad552/7265394
μ-PBWT, introducing a lightweight index for the PBWT data structure. It leverages the run-length encoding paradigm to significantly reduce the space requirements for solving two major problems: the SMEMs-finding (i.e. computing maximal matches) and SMEMs-location (i.e. finding occurrences).
μ-PBWT reduces the memory usage up to a factor of 20% compared to the best current PBWT-based indexing. In particular, μ-PBWT produces an index that stores high-coverage whole genome sequencing data of chromosome 20 in about a third of the space of its BCF file.

□ Local read haplotagging enables accurate long-read small variant calling
>> https://www.biorxiv.org/content/10.1101/2023.09.07.556731v1
An approximate haplotagging method that can locally haplotag long reads without having to generate variant calls. This approach uses local candidates to haplotag the reads and then the deep neural network model uses the haplotag approximation to generate high-quality variants.
This approach eliminates the requirement for having the first two steps for haplotagging the reads and reduces the overhead for extending support to newer platforms. Approximate haplotagging with candidate variants has comparable accuracy to haplotagging with WhatsHap.

□ BAGO: Bayesian optimization of separation gradients to maximize the performance of untargeted LC-MS
>> https://www.biorxiv.org/content/10.1101/2023.09.08.556930v1
BAGO, a Bayesian optimization method for autonomous and efficient LC gradient optimization. BAGO is an active learning strategy that discovers the optimal gradient using limited experimental data.
BAGO evaluates the retention of all detected features in an unbiased manner regardless of ion abundance and identity, providing a robust index representing global compound separation.
Multiple optimizations of general Bayesian optimization framework were applied to ensure the high efficiency of BAGO on a diverse range of gradient optimization problems.
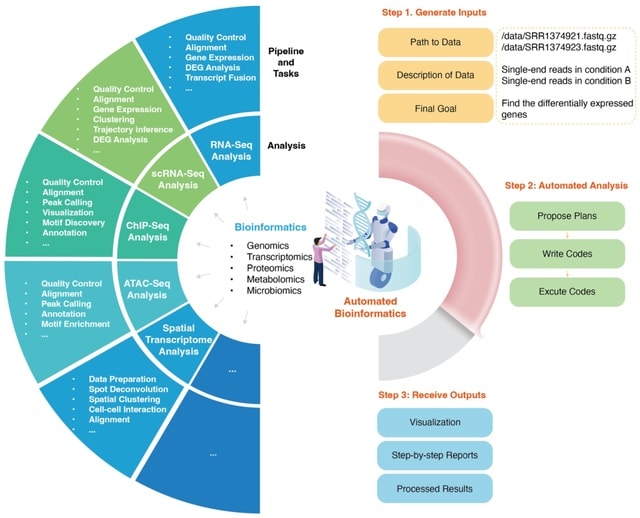
□ Automated Bioinformatics Analysis via AutoBA
>> https://www.biorxiv.org/content/10.1101/2023.09.08.556814v1
Auto Bioinformatics Analysis (AutoBA), the first autonomous AI agent meticulously crafted for conventional bioinformatics analysis. AutoBA streamlines user interactions by soliciting just three inputs: the data path, the data description, and the final objective.
AutoBA possesses the capability to autonomously generate analysis plans, write codes, execute codes, and perform subsequent data analysis. In essence, AutoBA marks the pioneering application of LLMs and automated AI agents in the realm of bioinformatics.

□ cloneRate: fast estimation of single-cell clonal dynamics using coalescent theory
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad561/7271182
cloneRate provides accessible methods for estimating the growth rate of clones. The input should either be an ultrametric phylogenetic tree with edge lengths corresponding to time, or a non-ultrametric phylogenetic tree with edge lengths corresponding to mutation counts.
This package provides the internal lengths and maximum likelihood methods for ultrametric trees and the shared mutations method for mutation-based trees. A fast way to simulate the coalescent (tree) of a sample from a birth-death branching process.

□ Hierarchical heuristic species delimitation under the multispecies coalescent model with migration
>> https://www.biorxiv.org/content/10.1101/2023.09.10.557025v1
Alternatively heuristic criteria based on population parameters under the MSC model (such as population/species divergence times, population sizes, and migration rates) estimated from genomic sequence data may be used to delimit species.
Extending the approach of species delimitation using the genealogical divergence index (gdi) to develop hierarchical merge and split algorithms for heuristic species delimitation, and implement them in a python pipeline called hhsd.

□ EvoDiff: Protein generation with evolutionary diffusion: sequence is all you need
>> https://www.biorxiv.org/content/10.1101/2023.09.11.556673v1
EvoDiff uses a discrete diffusion framework in which a forward process iteratively corrupts a protein sequence by changing its amino acid identities, and a learned reverse process, parameterized by a neural network, predicts the changes made at each iteration.
The reverse process can then be used to generate new protein sequences starting from random noise. EvoDiff's discrete diffusion formulation is mathematically distinct from continuous diffusion formulations previously used for protein structure design.
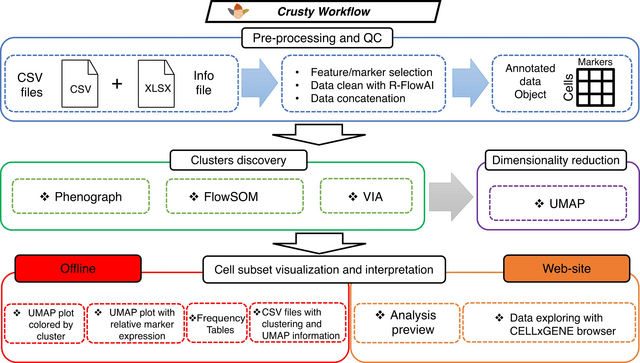
□ CRUSTY: a versatile web platform for the rapid analysis and visualization of high-dimensional flow cytometry data
>> https://www.nature.com/articles/s41467-023-40790-0
CRUSTY, an interactive, user-friendly webtool incorporating the most popular algorithms for FCM data analysis, and capable of visualizing graphical and tabular results and automatically generating publication-quality figures within minutes.

□ LIT: Identifying latent genetic interactions in genome-wide association studies using multiple traits
>> https://www.biorxiv.org/content/10.1101/2023.09.11.557155v1
LIT (Latent Interaction Testing) leverages multiple related traits for detecting latent genetic interactions. LIT is motivated by the observation that latent genetic interactions induce not only a differential variance pattern, but also a differential covariance pattern.
Combining the p-values from both approaches in aLIT maximized the number of discoveries while controlling the typeI error. LIT increased the power to detect latent genetic interactions compared to marginal testing, and the difference was drastic for certain genetic architectures.

□ The Interplay Between Sketching and Graph Generation Algorithms in Identifying Biologically Cohesive Cell-Populations in Single-Cell Data
>> https://www.biorxiv.org/content/10.1101/2023.09.15.557825v1
Combining a principled sketching approach with a simple k-nearest neighbor graph representation of the data can identify meaningful subsets of cells as robustly as, and sometimes better than, more sophisticated graph generation approaches.
Cell-similarity graphs are generally weighted, undirected, and simple. A weighted graph is one where each edge has a value assigned to it; large edge weights indicate strong connections between nodes.
Graph mining approaches perform better on sparse graphs than they do on dense graphs, and graph density varies significantly from the ultra-sparse GRASPEL to the 8-NN graph. Label propagation is more robust to noise and sparsity in the edges of a graph than Leiden clustering.










※コメント投稿者のブログIDはブログ作成者のみに通知されます