









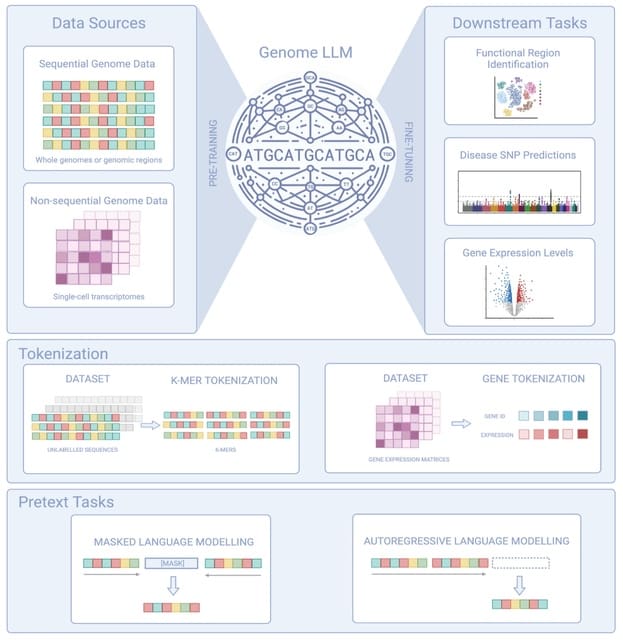

□ Genome LLM: To Transformers and Beyond: Large Language Models for the Genome
>> https://arxiv.org/abs/2311.07621
Genome LLMs, which are Transformer-hybrid models, are capable of processing both sequential and non-sequential data. It extracts signals to predict functional regions, identify disease-causing SNPs in individual DNA sequences, estimate gene expression, and more.
Genome LLMs can take in tokenized data. Another non-transformer genome LLM, HyenaDNA, achieves a context size of 1 million nucleotides, 500x larger than the largest of the foundational models utilizing full pairwise attention, the Nucleotide Transformer.

□ Universal Cell Embeddings: A Foundation Model for Cell Biology
>> https://www.biorxiv.org/content/10.1101/2023.11.28.568918v1
Universal Cell Embedding (UCE), a foundation model for single-cell gene expression. UCE is uniquely able to generate representations of new single-cell GE datasets with no model fine-tuning or retraining while still remaining robust to dataset and batch-specific artifacts.
UCE offers a unified biological latent space that can represent any cell, regardless of tissue or species. UCE generates an Integrated Mega-scale Atlas (IMA) of 36 million cells sampled from diverse biological conditions, demonstrating the emergent organization of UCE space.
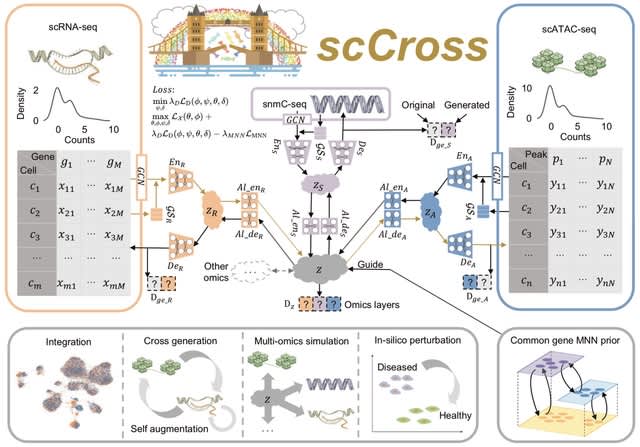
□ scCross: Bridging Modalities in Single–cell Multi–omics – Seamless Integration, Cross–modal Synthesis, and In–silico Exploration
>> https://www.biorxiv.org/content/10.1101/2023.11.22.568376v1
scCross employs a deep generative framework that combines the Variational Autoencoder (VAE) and Generative Adversarial Network (GAN) to adeptly integrate the Mutual Nearest Neighbors (MNN) technique for modality alignment.
The architecture of scCross operates on a two-step VAE to encode omics layers into a merged space. Inverting this methodology, any encoded data in this unified space can be reverted to any particular omics layer's latent representation using a dual-step decoding procedure.

□ HyGAnno: Hybrid graph neural network-based cell type annotation for single-cell ATAC sequencing data
>> https://www.biorxiv.org/content/10.1101/2023.11.29.569114v1
HyGAnno builds a hybrid graph by computing the similarity of gene expression and gene activity features b/n RNA cells & ATAC cells. ATAC cells showing similar gene-level similarity with RNA cell remain in the hybrid graph, whereas non-ATAC anchor cells are removed from the graph.
HyGAnno employs parallel graph neural networks to embed hybrid and ATAC graphs into separate latent spaces and minimizes the distance b/n the embeddings of the same ATAC anchor cells. This allows cell labels to be automatically transferred from scRNA-seq data to scATAC-seq data.
HyGAnno reconstructs a consolidated reference-target cell graph that shows more complex graph structures, thus inspiring us to describe ambiguous predictions based on abnormal target-reference cell connections.

□ Protein Design by Directed Evolution Guided by Large Language Models
>> https://www.biorxiv.org/content/10.1101/2023.11.28.568945v1
A general MLDE (machine learning-guided directed evolution) framework in which we apply recent advancements of Deep Learning in protein representation learning and protein property prediction to accelerate the searching and optimization processes.
ESM-2 adopts the encoder-only After that, the newly generated population Transformer architecture style with small modifications. The original Transformer uses absolute sinusoidal positional encoding to inform the model about token positions.
The ESM-2 model is capable of generating latent representations for individual amino acids inside a protein sequence. This is achieved through pre-training on a vast dataset consisting of millions of protein sequences including billions of amino acids.
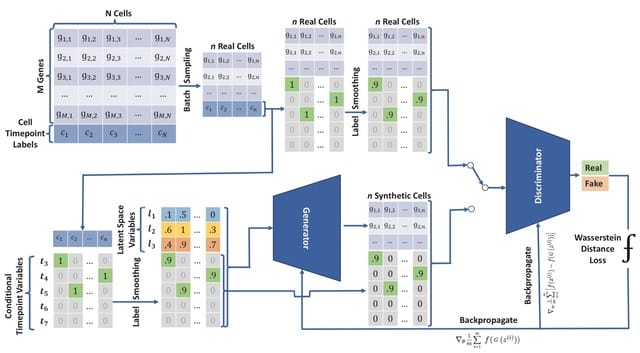
□ cwGAN: Hidden Knowledge Recovery from GAN-generated Single-cell RNA-seq Data
>> https://www.biorxiv.org/content/10.1101/2023.11.27.568840v1
cwGAN, a customized GAN method by incorporating the ideas of Conditional GAN and Wasserstein GAN with Gradient Prnalty using Label smoothing.
By formulating a quantitative score, Time-Point T-PCAVR (Time-Point PCA Variance Ratio) error, cwGAN can automatically select the most optimal GAN hyper-parameters. cwGAN preserves high-order relations by capturing cell developmental story as unknown semantic in the latent space.
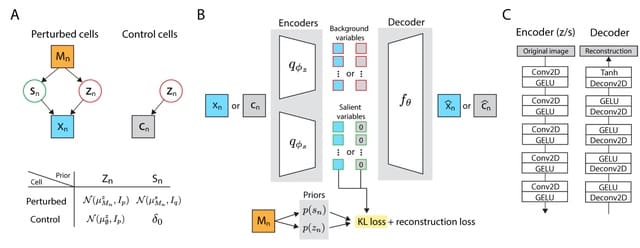
□ Multi-ContrastiveVAE disentangles perturbation effects in single cell images from optical pooled screens
>> https://www.biorxiv.org/content/10.1101/2023.11.28.569094v1
By analyzing a significant data set of over 30 million cells across more than 5, 000 genetic perturbations, Multi-Contrastive VAE automatically isolates multiple, intricate technical artifacts found in cell images without any prior information.
Multi-ContrastiveVAE (mcVAE) disentangles perturbation effects into separate latent spaces depending on whether the perturbation induces novel phenotypes unseen in the control cell population.
mcVAE can incorporate kernel-based independence measures to facilitate the enforcement of independence statements between the technical noise latent variables and the perturbation label.
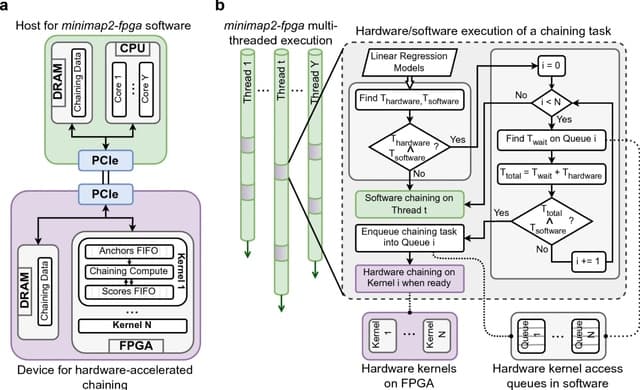
□ minimap2-fpga: Efficient end-to-end long-read sequence mapping using minimap2-fpga integrated with hardware accelerated chaining
>> https://www.nature.com/articles/s41598-023-47354-8
minimap2-fpga, a Field Programmable Gate Array (FPGA) based hardware-accelerated version of minimap2 that is end-to-end integrated. minimap2-fpga speeds up the mapping process by integrating an FPGA kernel optimised for chaining.
FPGA-based solutions include acceleration of the base-calling task in Oxford Nanopore sequence analysis, an integration of the GACT-X aligner architecture with minimap2, acceleration of minimap2’s chaining step and acceleration of selective genome sequencing.
For nanopore data, minimap2-fpga is 79% faster than minimap2 on the on-premise Intel FPGA system and 72% faster than minimap2 on the cloud Xilinx FPGA system when mapping without base-level alignment.
minimap2-fpga uses linear-regression based models to predict the time taken for each chaining task on hardware and software, allowing for more intelligent task-splitting decisions.
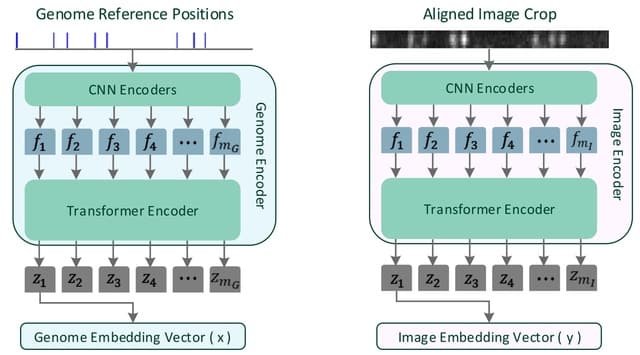
□ OM2Seq: Learning retrieval embeddings for optical genome mapping
>> https://www.biorxiv.org/content/10.1101/2023.11.20.567868v1
OM2seq is inspired by deep learning retrieval approaches, like Dense Passage Retrieval. The OM2Seq architecture takes cue from the Transformer-encoder utilized in the WavLM, featuring a convolutional feature encoder.
OM2Seq is trained on acquired OGM data to efficiently encode DNA fragment images and reference genome segments to a common embedding space, which can be indexed and efficiently queried using a vector database.
The OMSeq model is composed of 2 Transformer-encoders: one dubbed the Image Encoder, tasked with encoding DNA molecule images into embedding vectors, and another called the Genome Encoder, devoted to transforming genome sequence segments into their embedding vector counterparts.

□ scSemiProfiler: Advancing Large-scale Single-cell Studies through Semi-profiling with Deep Generative Models and Active Learning
>> https://www.biorxiv.org/content/10.1101/2023.11.20.567929v1
scSemiProfiler marries deep generative model with active learning strategies. This method adeptly infers single-cell profiles across large cohorts by fusing bulk sequencing data with targeted single-cell sequencing from a few carefully chosen representatives.
The core of the scSemiProfiler involves an innovative deep generative learning model. This model is engineered to intricately meld actual single-cell data profiles with the gathered bulk sequencing data, thereby capturing complex biological patterns and nuances.
scSemiProfiler uses a VAE-GAN architecture initially pretrained on single-cell sequencing data of selected representatives for self-reconstruction.
Subsequently, the VAE-GAN is further pretrained with a representative reconstruction bulk loss, aligning pseudobulk estimations from the reconstructed single-cell data with real pseudobulk.

□ vmrseq: Probabilistic Modeling of Single-cell Methylation Heterogeneity
>> https://www.biorxiv.org/content/10.1101/2023.11.20.567911v1
vmrseq is a novel computational tool developed for pinpointing variably methylated regions (VMRs) in scBS-seq data without prior knowledge on size or location.
High-throughput single-cell measurements of DNA methylation allows studying inter-cellular epigenetic heterogeneity, but this task faces the challenges of sparsity and noise. vmrseq overcomes these challenges and identifies variably methylated regions accurately and robustly.
vmrseq delineates the boundary of a VMR by removing any CpGs with estimates of hidden states uniform across the two groupings, effectively acting as a trimming step due to the assumption of at most one VMR per CR.
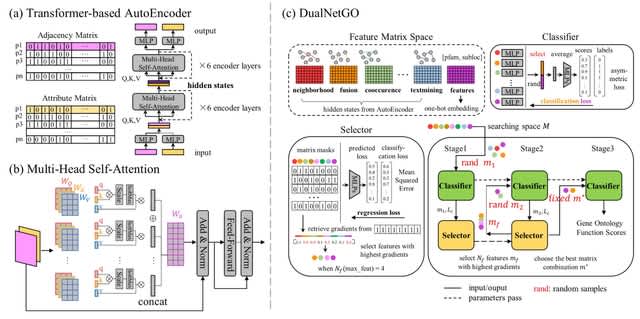
□ DualNetGO: A Dual Network Model for Protein Function Prediction via Effective Feature Selection
>> https://www.biorxiv.org/content/10.1101/2023.11.29.569192v1
DualNetGO is comprised of multilayer perceptron (MLP) components: a graph encoder for extracting graph information or generating graph embeddings and a predictor for predicting protein functions.
DualNetGO predicts protein function by effectively determining the combination of features from PPI networks and protein attributes without enumerating each possibility.
DualNetGO uses a feature matrix space that includes eight matrices: seven for graph embeddings of PPI networks from different evidence and one for protein domain and subcellular location.

□ MetaNorm: Incorporating Meta-analytic Priors into Normalization of NanoString nCounter Data
>> https://www.biorxiv.org/content/10.1101/2023.11.17.567577v1
MetaNorm, a Bayesian algorithm for normalizing NanoString nCounter GE data. MetaNorm is based on RCRnorm, a method designed under an integrated series of hierarchical models that allow various sources of error to be explained by different types of probes in the nCounter system.
MetaNorm employs priors carefully constructed from a rigorous meta-analysis to leverage information from large public data. MetaNorm improves RCRnorm by yielding more stable estimation of normalized values, better convergence diagnostics and superior computational efficiency.
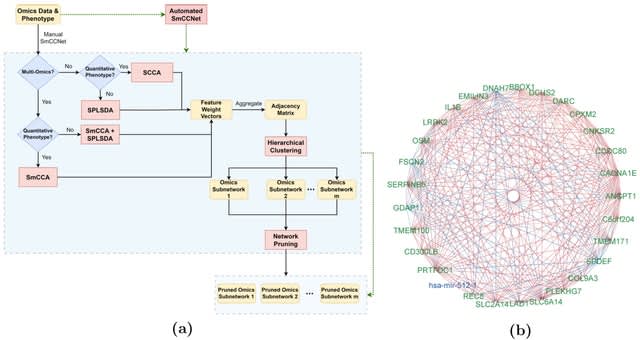
□ SmCCNet 2.0: an Upgraded R package for Multi-omics Network Inference
>> https://www.biorxiv.org/content/10.1101/2023.11.20.567893v1
SmCCNet (Sparse multiple Canonical Correlation Network Analysis) is a canonical correlation-based integration method that reconstructs phenotype-specific multi-omics networks. SmCCNet 2.0 incorporates numerous new features including generalization to single or multi-omics data.
SmCCNet 2.0 uses a novel stepwise hybrid approach is developed for multi-omics data with a binary phenotype by filtering molecular features to identify interconnected molecular features, then implementing Sparse Partial Least Squared Discriminant Analysis.
□ RF-PHATE: Gaining Biological Insights through Supervised Data Visualization
>> https://www.biorxiv.org/content/10.1101/2023.11.22.568384v1
RF-PHATE combines Random Forest geometry- and accuracy-preserving proximities, with the Dimensionality Reduction method PHATE to visualize the inherent structure of the features that are relevant to the supervised task while ignoring the irrelevant features.
PHATE uses von Neumann Entropy (VNE) of the diffused operator. RF-PHATE is able to ignore irrelevant features and capture the true structure of the artificial tree data. They used Dynamic Time Warping as a proximity measure.
The proximities are row-normalized, and damping is applied to form the diffusion probabilities, which are stored in a Markov transition matrix. The global relationships are learned by diffusion, which is equivalent to simulating all possible random walks.
□ LncPNdeep: A long non-coding RNA classifier based on Large Language Model with peptide and nucleotide embedding
>> https://www.biorxiv.org/content/10.1101/2023.11.29.569323v1
LncPNdeep incorporates both peptide and nucleotide embedding from masked language modeling (MLM), being able to discover complex associations between sequence information and lncRNA classification.
LncPNdeep utilized the Bigbird, Longformer, and ProteinTrans models for the embedding’s extraction. However, other Masked Language Models such as ProteinBERT and DNABERT remain to be assessed for potential improvement in LncPNdeep.

□ Tensor categories
>> https://arxiv.org/abs/2311.05789
A tensor category is finite if all hom-spaces are finite dimensional and any object has a finite length (filtration w/ simple factors). As an abelian category a finite tensor category is equivalent to the category of finite dimensional modules over a finite dimensional algebra.
As the result a finite tensor category is finitely complete and cocomplete, and a tensor functor between finite tensor categories has left and right adjoints. In particular, internal action homs for a finite module category exist.
Concepts crucial for the emergent theory of tensor categories came from or play an important role in: non-degenerate braided fusion categories, module categories, Witt equivalence. Higher categorical analogues of tensor categories play an important role in 4d topological field.

□ Community Detection with the Map Equation and Infomap: Theory and Applications
>>
Infomap is a greedy stochastic search algorithm designed to minimize the map equation and detect two-level and multilevel flow communities in networks.
The Infomap search algorithm is inspired by the Louvain algorithm for modularity maximization but uses additional fine-tuning and coarse-tuning steps, similar to how the Leiden algorithm later refined Louvain.
The multilevel phase aims to reduce the codelength by adding further index levels to a two-level partition. It contains two stages.
In stage 1, Infomap compresses inter-module transitions by first aggregating the network at the module level. This creates a network where nodes represent the previous modules, and inter-module links are merged.
Second, Infomap uses the two-level algorithm to partition the aggregated network. The resulting two-level partition comprises a three-level partition when interpreted in the context of the network before aggregation.
Infomap repeats stage 1 as long as aggregating and partitioning the network and adding one more index level per iteration yields a non-trivial solution.

□ CGCom: a framework for inferring Cell-cell Communication based on Graph Neural Network
>> https://www.biorxiv.org/content/10.1101/2023.11.10.566642v1
CGCom models cell-to-cell relationships and the intricate communication patterns. The framework takes as input a series directed sub-graph generated from cell physical locations, combined with ligand expression values, and utilizes cell type information as the training objective.
The paired cell communication coefficient is computed from the attention scores in the well-trained Graph Attention Network (GAT graph) classifier. CGCom then introduces a heuristic computational algorithm to quantify communication between neighboring cells through various ligand-receptor pairs.
CGCom outperforms multilayer perceptron (MLP) baseline. It employs the attention scores from GAT classifier to infer cell communication on the same datasets, revealing common communication patterns between the three datasets.
CGCom takes the GE matrix. The GAT learns the ligand expression patterns of different cell types in a semi-supervised model. It extracts the attention score in each graph embedding layer in the GAT from the trained model and infer the communication using a heuristic rule.

□ SQUID: Interpreting cis-regulatory mechanisms from genomic deep neural networks using surrogate models
>> https://www.biorxiv.org/content/10.1101/2023.11.14.567120v1
SQUID (Surrogate Quantitative Interpretability for Deepnets), an interpretability framework for genomic DNNs that overcomes these limitations. SQUID uses surrogate models with interpretable parameters-to approximate the DNN function within localized regions of sequence space.
SQUID applies MAVE-NN, a quantitative modeling framework developed for analyzing multiplex assays of variant effects (MAVEs), to in silico MAVE datasets generated using the DNN as an oracle.
SQUID models DNN predictions in a user-specified region of sequence space, accounts for the nonlinearities and heteroscedastic noise present in DNN predictions, and (optionally) quantifies specific epistatic interactions.

□ scReadSim: a single-cell RNA-seq and ATAC-seq read simulator
>> https://www.nature.com/articles/s41467-023-43162-w
scReadSim, a single-cell RNA-seq and ATAC-seq read simulator that allows user-specified ground truths and generates synthetic sequencing reads by mimicking real data. At both read-sequence and read-count levels, scReadSim mimics real scRNA-seq and scATAC-seq data.
scReadSim mimics real data by first generating realistic UMI counts and then simulating reads. The synthetic UMI count matrix serves as the ground truth for benchmarking scRNA-seq UMI deduplication tools which all process reads into a UMI count matrix.
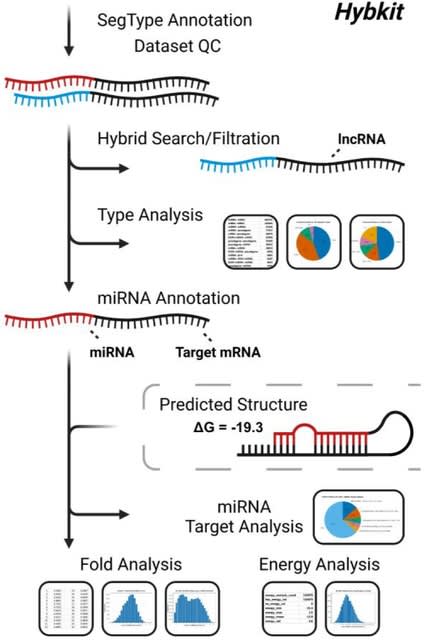
□ Hybkit: a Python API and command-line toolkit for hybrid sequence data from chimeric RNA methods
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad721/7451011
Hybkit enables the flexible classification and annotation of identified hybrid segments, identification of miRNA-containing hybrids, and filtration of records based on sequence identifiers and other annotation information.
Built-in plotting features allow visualization of analysis results, including plotting the distributions of segment types and miRNA targets. Hybkit can merge information from hyb files with corresponding predicted molecular secondary structure ("fold") files in the Vienna format.
Hybkit provides insight into potential miRNA/target affinity and functionality of miRNA/target interactions. Hybkit additionally provides a file-format specification for "hyb" files for standardized file parsing and annotation.
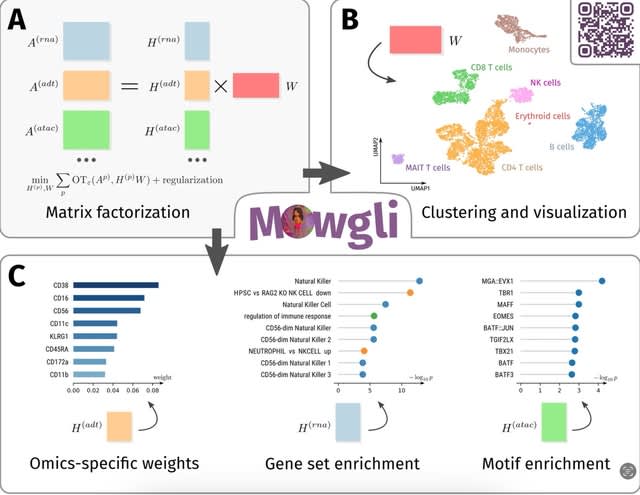
□ Mowgli: Paired single-cell multi-omics data integration
>> https://www.nature.com/articles/s41467-023-43019-2
Mowgli (Multi-Omics Wasserstein inteGrative anaLysIs), a novel method for the integration of paired multi-omics data with any type and number of omics, combining integrative Nonnegative Matrix Factorization and Optimal Transport.
Mowgli employs integrative NMF, and contains omics-specific weights for each latent dimension, which can be used for the biological characterization of the latent dimensions through gene set enrichment or motif enrichment analysis.
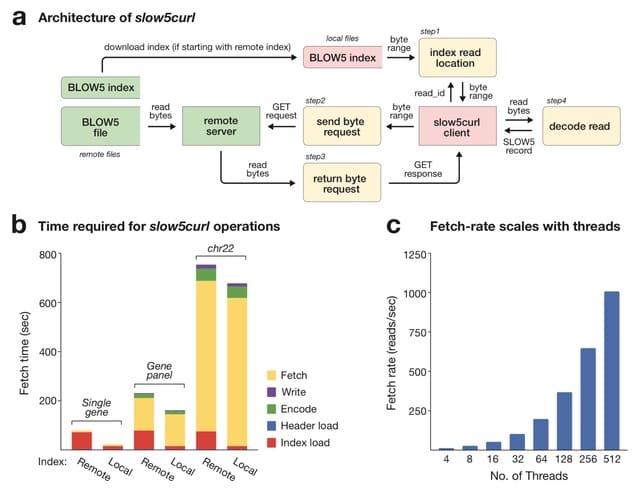
□ slow5curl: Streamlining remote nanopore data access
>> https://www.biorxiv.org/content/10.1101/2023.11.28.569128v1
slow5curl, a simple command line tool and underlying software library to improve remote access to nanopore signal datasets. Slow5curl enables a user to extract and download a specific read or set of reads from a dataset on a remote server, avoiding the need to download the entire file.
Slow5curl uses highly parallelised data access requests to maximise speed. slow5curl can facilitate targeted reanalysis of remote nanopore cohort data, effectively removing data access as a consideration.

□ PMFFRC: a large-scale genomic short reads compression optimizer via memory modeling and redundant clustering
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05566-9
PMFFRC (Parallel Multi-FastQ-Files Reads Clustering) performs joint clustering compression on the Reads in multiple FastQ files by modeling the system memory, the peak memory overhead of the cascading compressor, the numeral of files, and the numeral of sequencing.
PMFFRC initiates the analysis from the matrix element with the highest similarity score and employs a straightforward "first cluster first priority" principle when clustering fastq files.
The FastqCLS compressor incorporates the ZPAQ algorithm, which employs context modelling and arithmetic coding. This enables FastqCLS to detect patterns and character dependencies in the reads, utilizing context models and exploiting redundancy at the nucleotide character level.
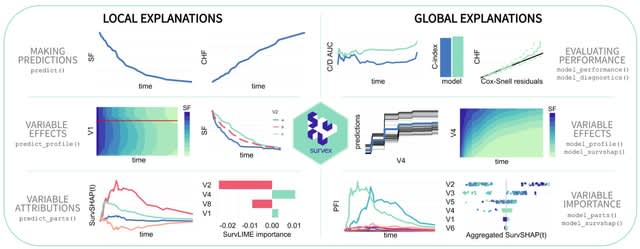
□ survex: an R package for explaining machine learning survival models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad723/7457480
survex provides model-agnostic explanations for machine learning survival models. It is based on the DALEX and iml, which offer a diverse spectrum of XAI techniques. XAI techniques. Their core focus remains rooted in the domain of explaining classification and regression models.
survex enables the assessment of model reliability and the detection of biases. survex offers specifically tailored explanations that incorporate the time dimension inherent in the survival models' predictions.
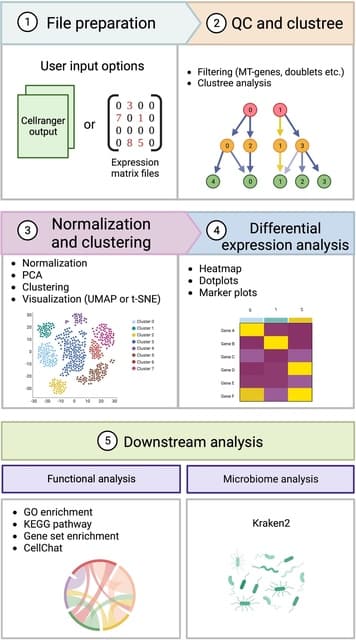
□ Cellsnake: a user-friendly tool for single-cell RNA sequencing analysis
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad091/7330891
cellsnake can utilize different scRNA-seq algorithms to simplify tasks such as automatic mitochondrial (MT) gene trimming, selection of optimal clustering resolution, doublet filtering, visualization of marker genes, enrichment analysis, and pathway analysis.
Cellsnake allows parallelization and readily utilizes HPC platforms. Cellsnake provides metagenome analysis if unmapped reads are available. Cellsnake generate intermediate files that can be stored, extracted, shared, or used later for more advanced analyses.

□ A PhyloFisher utility for nucleotide-based phylogenomic matrix construction; nucl_matrix_constructor.py
>> https://www.biorxiv.org/content/10.1101/2023.11.30.569490v1
PhyloFisher currently includes a manually curated starting dataset of 240 proteins from 304 eukaryotic taxa representing the full breadth of known diversity in the eukaryotic tree of life.
Importantly, this dataset also includes identified paralogs of each of the 240 proteins from all investigated taxa which is crucial for the identification of probable orthologs.
nucl_matrix_constructor.py, an expansion of the PhyloFisher starting DB, and an update to PhyloFisher that maintains DNA sequences. It takes the output of prep final dataset, which contains amino acid sequences for each gene, and a TSV w/ paths to coding sequence files as input.
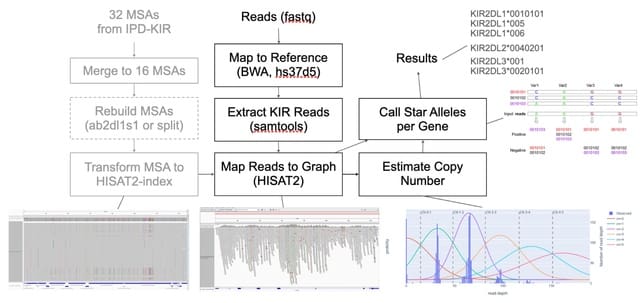
□ Graph-KIR: Graph-based KIR Copy Number Estimation and Allele Calling Using Short-read Sequencing Data
>> https://www.biorxiv.org/content/10.1101/2023.11.29.568665v1
Graph-KIR aims to estimate the copy number of genes and calls full-resolution (7 digits) KIR alleles from a whole genome sequencing sample. Graph-KIRvis capable of independently typing KIR alleles per sample with no reliance on the distribution of any framework gene in a cohort.
Graph-KIR utilizes HISAT2, a graph read mapper, to map short reads to custom-built indexes. The highly accurate graph mapping enables Graph-KIR to estimate copy number per sample independently, thanks to the higher linearity b/n copy number and read depth in the graph alignment.
□ Wavelet—Graph変換とDynamic Time Warpingを用いた遺伝子発現クラスタリング










※コメント投稿者のブログIDはブログ作成者のみに通知されます