









□ Loop Assembly: a simple and open system for recursive fabrication of DNA circuits:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/15/247593.full.pdf
The vectors contain modular sites for hybrid assembly using sequence overlap methods. Loop assembly provides a simple generalised solution for DNA construction with standardised parts. Such approaches would make the DNA fabrication process host-agnostic, promoting the development of universal DNA assembly systems using standards such as the common syntax, which would provide unprecedented exchange of DNA components within the biological sciences. This is due to the layers of abstraction provided by the use of a common syntax for DNA elements, the use of a simple scheme of plasmid vectors during assembly, common laboratory procedures & reagents, and streamlined protocols for the design & set up of Loop assembly reactions.
□ Highly parallel direct RNA sequencing on an array of nanopores:
>> https://www.nature.com/articles/nmeth.4577
nanopore direct RNA-seq, a highly parallel, real-time, single-molecule method that circumvents reverse transcription or amplification steps.

□ CWL-Airflow: a lightweight pipeline manager supporting Common Workflow Language:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/17/249243.full.pdf

□ MoNaLISA: Enhanced photon collection enables four dimensional fluorescence nanoscopy of living systems:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/17/248880.full.pdf
By maximizing the detected photon flux MoNaLISA enables prolonged (40-50 frames) and large (50 x 50 um2) recordings at 0.3-1.3 Hz with enhanced optical sectioning ability.
□ Observation weights to unlock bulk RNA-seq tools for zero inflation and single-cell applications:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/18/250126.full.pdf
a weighting strategy, based on a zero-inflated negative binomial (ZINB) model, that identifies excess zero counts and generates gene and cell-specific weights to unlock bulk RNA-seq DE pipelines for zero-inflated data, boosting performance for scRNA-seq.
□ Download new Supernova 2.0 sample datasets for humans, plants, insects and other animals, that showcase improvements such as longer contigs, scaffolds, and phase blocks. #LinkedReads
>> http://bit.ly/2EVvosa
□ GFA output from Falcon genome assembly can be input to Bandage (https://github.com/rrwick/Bandage ) for visualization @PacBio #SMRTBFX
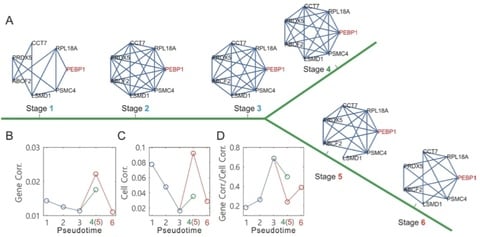
□ Topographer Reveals Stochastic Dynamics of Cell Fate Decisions from Single-Cell RNA-Seq Data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/21/251207.full.pdf
Topographer, a bioinformatics pipeline that can construct an intuitive developmental landscape where by ‘intuitive’ mean that every cell is equipped with both potential and pseudotime, quantify stochastic dynamics of cell types by estimating both their fate probabilities and transition probabilities among them, and infer dynamic characteristics of transcriptional bursting kinetics along the developmental trajectory.
□ empiricIST: The fitness landscape of the codon space across environments:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/23/252395.full.pdf
Apart from its main program – the Bayesian MCMC program – empiricIST provides Python and shell scripts for data pre- and post-processing. A structural analysis indicates that synonymous effects can be mediated by changes in mRNA stability and variation in codon preference. However, effects are strongly dependent on the residue position under study, which makes a clear identification of the predictors of synonymous effects difficult. Overall, this study demonstrates how synonymous mutations can directly impact both the path and endpoint of an adaptive walk, and thus highlights the importance of their consideration.
□ Harmonizing semantic annotations for computational models in biology:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/23/246470.full.pdf
A common annotation protocol is also a critical component of model composition. Applying the protocol, model-merging tools such as SemGen can recognize the biological commonalities and then use that information to compare the models’ biological content and guide their assembly. Without a harmonized approach to semantic annotation, modelers would be required to manually identify the biological overlap between models.

□ TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding:
>> https://www.nature.com/articles/nmeth.4582
TimeLapse-seq, which uses oxidative-nucleophilic-aromatic substitution to convert 4-thiouridine into cytidine analogs, yielding apparent U-to-C mutations that mark new transcripts upon sequencing. TimeLapse-seq is a single-molecule approach that is adaptable to many applications and reveals RNA dynamics and induced differential expression concealed in traditional RNA-seq.

□ Automatic error control during forward flux sampling of rare events in master equation models:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/27/254896.full.pdf
Complex, nonequilibrium regulatory and signaling networks connect these molecules through positive and negative interactions, naturally resulting in a large number of metastable states within the space, representing different cellular phenotypes. Even with oversampling to control land- scape error, speedups on the order of 100X for systems with long first passage times can be expected. Higher dimensional systems have additional sources of error and the extra error can be traced to correlations between phases due to roughness on the probability landscape.
□ Markov Katana: a Novel Method for Bayesian Resampling of Parameter Space Applied to Phylogenetic Trees
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/24/250951.full.pdf
the Markov katana bootstrapping approach to phylogenetic tree searching can be a highly effective means for finding Bayesian posterior topologies and branches. Many previous phylogenetic tree-search methods use the provided sequences for only the likelihood calculations, but Markov katana introduces a new way to explore tree space informed by the sequences.

□ COSSMO: Predicting Competitive Alternative Splice Site Selection using Deep Learning:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/29/255257.full.pdf
COSSMO is able to predict the most frequently used splice site with an accuracy of 70% on unseen test data, which compares to only around 35% accuracy for MaxEntScan. COSSMO vastly outperforms MaxEntScan as well on negative cross-entropy, meaning COSSMO is not only more likely to predict the correct dominant splice site but will also fit the PSI distribution better overall.

□ LiMMBo: a simple, scalable approach for linear mixed models in high-dimensional genetic association studies:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/30/255497.full.pdf
LiMMBo enables multivariate analysis of high-dimensional phenotypes based on linear mixed models with bootstrapping. It builds on and can be used in combination with Limix framework. The majority of compute time is needed for the variance decomposition of the bootstrapped subsets, which can be trivially parallelised across bootstraps. the time taken by the standard REML approach quickly exceeds the time of LiMMBo & becomes infeasible for more than 30 traits. By fitting the bootstrap average to the closest true covariance, LiMMBo ensures positive-semidefiniteness of the covariance while avoiding ill-conditioned matrices, which usually introduces large biases in the final use of these models.

□ DrAnneCarpenter:
Look what deep learning can do for blood stored at blood banks! Our preprint just out: "Label-free assessment of red blood cell storage lesions by deep learning"
>> https://www.biorxiv.org/content/early/2018/01/30/256180
□ Rapid multiplex small DNA sequencing on the MinION nanopore sequencing platform
>> https://www.biorxiv.org/content/early/2018/01/31/257196
an ultra-rapid multiplex library preparation and sequencing method for the MinION is presented and applied to accurately test normal diploid and aneuploidy samples' genomic DNA in under three hours,
□ ANIMA: Association Network Integration for Multiscale Analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/31/257642.full.pdf
Meta-analysis of multiple related expression datasets can lead to insights not available from analysis of any single datasets, and can highlight common patterns of transcript abundance across different conditions, or meaningful differences across highly common conditions. A second approach to meta-analysis was implemented using virtual cells based on WGCNA modules. Application of the two meta-analysis approaches allows comparison of arbitrary datasets to detect similarities and differences at modular and cellular levels.
□ CALISTA: Clustering And Lineage Inference in Single-Cell Transcriptional Analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/01/31/257550.full.pdf
the pseudotimes are normalized to have values between 0 and 1 by dividing with the maximum possible value among the clusters. The user can manually assign the pseudotimes of the clusters based on some prior knowledge. Subsequently, each cell is assign to one of the state transition edges that are incident to the cluster containing the cell using the maximum likelihood principle, and then given a pseudotime that corresponds to the maximum point. Finally, given a developmental path in the linage progression, CALISTA can generates a pseudotemporal ordering of cells belonging to the state transition edges in the defined path.
□ ARGs-OAP v2.0 with an Expanded SARG Database and Hidden Markov Models for Enhancement Characterization and Quantification of Antibiotic Resistance Genes in Environmental Metagenomes.
>> http://dlvr.it/QFPNvY
□ A tutorial on Bayesian parameter inference for dynamic energy budget models
>> https://www.biorxiv.org/content/early/2018/02/05/259705
Dynamic energy budget (DEB) theory provides compact models to describe the acquisition and allocation of organisms over their full life cycle of bioenergetics.
□ ASGAL: Aligning RNA-Seq Data to a Splicing Graph to Detect Novel Alternative Splicing Events:
>> https://www.biorxiv.org/content/biorxiv/early/2018/02/07/260372.full.pdf
ASGAL is the first tool specifically designed for mapping RNA-Seq data directly to a splicing graph. Differently from SplAdder, which enriches a splicing graph representing the gene annotation using the splicing information contained in the input spliced alignments, and then analyzes this enriched graph to detect the AS events differentially expressed in the input samples, ASGAL directly aligns the input sample to the splicing graph of the gene of interest and then detects the AS events which are novel with respect to the input gene annotation, comparing the obtained alignments with it.
□ RNentropy: an entropy-based tool for the detection of significant variation of gene expression across multiple RNA-Seq experiments
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gky055/4829696
Other large-scale studies on human gene expression highlighted individual variability, also correlating it with eQTLs: RNentropy however permits to study this phenomenon more in depth, with a detailed and simultaneous individual- or tissue-based assessment and classification of the specificity of the expression of each single gene.

□ Reactome graph database: Efficient access to complex pathway data:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005968
The relational database is converted to a graph database via the batch importer that relies on the Domain Model. Spring Data Neo4j and AspectJ are two main pillars for the graph-core, which also rests on the Domain Model. Users access services or use tools that make direct use of the graph-core as a library that eliminates the code boilerplate for data retrieval and offers a data persistency mechanism. Finally, export tools take advantage of Cypher to generate flat mapping files.
□ KrakenHLL: Confident and fast metagenomics classification using unique k-mer counts:
>> https://www.biorxiv.org/content/biorxiv/early/2018/02/09/262956.full.pdf
KrakenHLL is based on the ultra-fast classification engine Kraken and combines it with HyperLogLog cardinality estimators. The main idea behind the method is that long runs of leading zeros are unlikely in random hashes. E. g., it’s expected to see every fourth hash start with one 0-bit before the first 1-bit (01 2), and every 32nd hash starts with 00001 2.
□ RamDA-seq using BioJulia: Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs
>> https://www.nature.com/articles/s41467-018-02866-0
random displacement amplification sequencing (RamDA-seq), the first full-length total RNA-sequencing method for single cells. RamDA-seq shows high sensitivity to non-poly(A) RNA and near-complete full-length transcript coverage.

□ A maximum-entropy model for predicting chromatin contacts:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005956
if the maximum entropy model is reliably capturing the essential aspects that connect sequence to structure and if one believes that these aspects are conserved across different cell types, one could use a fitted model from one cell type to predict sequence in another for which only structural information is known. a first-order maximum-entropy model only constrained to reproduceone-spin statistics(σk)of the experimental distributions of neighborhoods~σ. Similarly to the second-order model, the first-order maximum-entropydistribution can be derived using the method of Lagrange multipliers.
□ mason_lab:
@10xgenomics has 3 new products: single cell ATAC-seq, single cell CNV mapping for clonal evolution, and myriad single cell feature barcodes #AGBT18 AND an updated platform for Cell-Beads Gel-Beads (CBGBs), works on current Chromium, w/ protein digestion & alkaline denaturation.
>> https://www.10xgenomics.com/future/

□ A Hierarchical Anti-Hebbian Network Model for the Formation of Spatial Cells in Three-Dimensional Space:
>> https://www.biorxiv.org/content/biorxiv/early/2018/02/13/264366.full.pdf
Place and grid representationsare high level spatial encoding produced by combing the high variance-high dimensional sensory information. Learning rules of the anti-hebbian network performs PCA like transformation i.e. projecting the high dimensional spatial inputs provided by the path integration oscillators to those weight vectors in the direction of maximal variance.
□ MasterPATH: network analysis of functional genomics screening data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/02/13/264119.full.pdf
MasterPATH extracts subnetwork built from the shortest paths between hit genes to so called “final implementers” - genes that are involved in molecular events responsible for final phenotypical realization (if known) or between hit genes (if “final implementers” are not known). The method calculates centrality score for each node and each linear path in the subnetwork as the number of paths found in the previous step that pass through the node and the linear path.

□ An adaptive configuration interaction approach for strongly correlated electrons with tunable accuracy:
>> http://aip.scitation.org/doi/10.1063/1.4948308
The exponential scaling of the number of determinants with respect to the number of orbitals required for FCI calculations prevents its use for all but trivially small systems, or for active space calculations no larger than 18 electrons in 18 orbitals. Recently, the density matrix renormalization group, and stochastic CI approaches such as Monte Carlo CI and FCI Quantum Monte Carlo have risen as promising alternatives to FCI and complete active space CI (CASCI), allowing for the description of chemically interesting systems.










※コメント投稿者のブログIDはブログ作成者のみに通知されます