









□ Trajan: Dynamic pseudo-time warping of complex single-cell trajectories:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/17/522672.full.pdf
Trajan, a novel method that allows for the first time the alignment of complex (non-linear) single-cell trajectories. Trajan automatically pairs similar biological processes between conditions and aligns them in a globally consistent manner. Trajan identifies and aligns the core paths without prior information, and does not make any assumptions concerning the algorithm used to reconstruct the trajectory and can in principle be coupled with any available reconstruction method.
Originally introduced to compare phylogenetic trees, in Trajan adopting arboreal matchings to perform an unbiased alignment enabling the meaningful comparison of gene expression dynamics along a common pseudo-time scale.
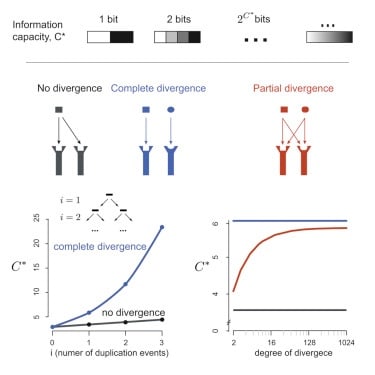
□ The Limited Information Capacity of Cross-Reactive Sensors Drives the Evolutionary Expansion of Signaling:
>> https://www.cell.com/cell-systems/fulltext/S2405-4712(18)30480-0
Using information theory, augmentation of capacity by an increase in the copy number is strongly limited by logarithmic diminishing returns, and counter to conventional biochemical wisdom, refinements of the response mechanism do not increase the overall signaling capacity. the expansion of signaling components via duplication and enlistment of promiscuously acting cues is virtually the only accessible evolutionary strategy to achieve overall high-signaling capacity despite overlapping specificities.

□ DIABLO: an integrative approach for identifying key molecular drivers from multi-omic assays:
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/bty1054/5292387
DIABLO, a multi-omics integrative method that seeks for common information across different data types through the selection of a subset of molecular features, while discriminating between multiple phenotypic groups. DIABLO is versatile, allowing for modular-based analyses and cross-over study designs.

>> https://store.nanoporetech.com/minion-mk1c-basic-starter-pack.html/
The MinION Mk1c combines the real-time, rapid, portable sequencing of MinION and Flongle with real-time, powerful GPU based computing and a high resolution screen.
□ The MinION Mk1c combines the real-time, rapid, portable sequencing of MinION and Flongle with real-time, powerful GPU based computing and a high resolution screen. Pre-order in the store: http://bit.ly/2MuaA09 or register your interest: http://bit.ly/2MrTmAB
□ Dna-nn: Model and predict short DNA sequence features with neural networks
>> https://github.com/lh3/dna-nn
Dna-nn implements a proof-of-concept deep-learning model to learn relatively simple features on DNA sequences. So far it has been trained to identify the (ATTCC)n and alpha satellite repeats, which occupy over 5% of the human genome. Taking the RepeatMasker annotations as the ground truth, dna-nn is able to achieve high classification accuracy.

□ On the Complexity of Sequence to Graph Alignment:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/17/522912.full.pdf
when changes are permitted in graphs either standalone or in conjunction with changes in the query, the sequence to graph alignment problem is NP-complete under both Hamming and edit distance models for alphabets of size ≥ 2. For the case where only changes to the sequence are permitted, present an O(|V | + m|E|) time algorithm, where m denotes the query size, and V and E denote the vertex and edge sets of the graph, respectively.
□ Bioshake: a Haskell Embedded Domain Specific Language (EDSL) for bioinformatics pipelines:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/24/529479.full.pdf
Bioshake abstracts execution so that jobs can either be executed directly or submitted to a cluster. s the main data type is recursively defined, outputs of a stage can always be referenced by subsequent stages without explicit non-linear constructs. the alignments used for variant calling are available for a subsequent variant annotation stage without explicitly introducing non-linearity.
Bioshake EDSL provides the EDAM ontology in the EDAM module. This module provides EDAM terms identified by their short name, along with some template Haskell for associating EDAM terms to types.
□ Causal Mediation Analysis Leveraging Multiple Types of Summary Statistics Data:
>> https://arxiv.org/pdf/1901.08540.pdf
This Bayesian approach to summary-based analysis truly seeks to answer causal questions, explicitly constructing proxy- variables to capture the unmediated and unwanted effects. Moreover, this framework can robustly work against high- dimensionality and collinearity of the parametric space, naturally induced by human genetics.

□ A pitfall for machine learning methods aiming to predict across cell types:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/04/512434.full.pdf
a natural strategy is to train the model on experimental data from some cell types and evaluate performance on one or more held-out cell types. A more sophisticated strategy would be to use as a baseline the activity of a cell type in the training set that is empirically similar to the target cell type.

□ IMCC: Quantitative Analysis of the Inter-module Connectivity for Bio-network:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/10/516237.full.pdf
IMCC deciphered characteristic pathological “core-periphery” structure of modular map and functional coordination module pair.
□ TOPAS: network-based structural alignment of RNA sequences:
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz001/5284906
TOPAS first constructs a topological network based on the predicted folding structure, which consists of sequential edges and structural edges weighted by the base-pairing probabilities. The computational complexity of TOPAS is significantly lower than that of the Sankoff-style dynamic programming approach, while yielding favorable alignment results, and has advantage in capability of handling RNAs with pseudoknots while predicting the RNA structural alignment.

□ Memory-driven computing accelerates genomic data processing:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/13/519579.full.pdf
memory-driven computing (MDC), a novel memory-centric hardware architecture, is an attractive alternative to current processor-centric compute infrastructures. Adapting transcriptome assembly pipelines for MDC reduced compute time by 5.9-fold for the first step. pseudoalignment by near-optimal probabilistic RNA-seq quantification (kallisto) was accelerated by more than two orders of magnitude with identical accuracy and indicated 66% reduced energy consumption. One billion RNA-seq reads were processed in just 92 seconds.

□ MS-Helios: a Circos wrapper to visualize multi-omic datasets:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2564-9
MS-Helios enables users to build circular plots with a non-genomic basis for exploration of high-dimensional multi-omic data without requiring any prior knowledge with Circos.
□ Highly-accurate long-read sequencing improves variant detection and assembly of a human genome:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/13/519025.full.pdf
a protocol based on single-molecule, circular consensus sequencing (CCS) to generate highly-accurate (99.8%) long reads averaging 13.5 kb and applied it to sequence the well-characterized human HG002/NA24385. CCS reads achieving precision and recall above 99.91% for SNVs, 95.98% for indels, and 95.99% for structural variants.

□ BEHST: genomic set enrichment analysis enhanced through integration of chromatin long-range interactions:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/15/168427.full.pdf
Biological Enrichment of Hidden Sequence Targets (BEHST), finds Gene Ontology (GO) terms enriched in genomic regions more precisely and accurately than existing methods. BEHST incorporates experimental evidence of these interactions from Hi-C datasets. These datasets include chromatin loops that bring linearly distal regions up to hundreds of kilobases away within spatial proximity.
□ MACHINE LEARNING AND THE CONTINUUM HYPOTHESIS:
>> https://arxiv.org/pdf/1901.04773.pdf
point out that part of the set-theoretic machinery is related to a result of Kuratowski about decompositions of finite powers of sets and show that there is no Borel measurable monotone compression function on the unit interval. exhibiting an abstract machine-learning situation where the learnability is actually neither provable nor refutable on the basis of the axioms of ZFC.
□ PaKman: Scalable Assembly of Large Genomes on Distributed Memory Machines:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/17/523068.full.pdf
using a deterministic algorithm to “wire” the paths through each vertex in a PaK-Graph to enable non-redundant walks without further coordination. This approach reduces the often complicated implementation of the walk phase into an embarrasingly parallel procedure.
□ Minerva: an alignment- and reference-free approach to deconvolve Linked-Reads for metagenomics:
>> https://genome.cshlp.org/content/29/1/116
□ PRINCESS: Framework for Comprehensive Detection and Phasing of SNP and SVs:
>> https://figshare.com/articles/SMRTInformatics2019/7597577
PRINCESS (Phase Resolution IN Comprehensive Small and Structural variants) will provide Parameter optimization, Provides QC and automatic parameter adaption and Optional takes parental SNPs into account. PRINCESS using Snakemake pipeline to CallSNPs, CallSVs and Phase SNPs + SVs. PRINCESS combines methods from Clairvoyante, NGMLR, Sniffles, and HapCut2.

□ SpliceAI: Splicing from Primary Sequence with Deep Learning:
>> https://www.cell.com/cell/fulltext/S0092-8674(18)31629-5
SpliceAI is 32-layer a deep neural network that accurately predicts splice junctions from an arbitrary pre-mRNA transcript sequence, enabling precise prediction of noncoding genetic variants that cause cryptic splicing.
□ VPAC: Variational projection for accurate clustering of single-cell transcriptomic data:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/18/523993.full.pdf
for accurate clustering of single-cell transcriptomic data through variational projection, which assumes that single-cell samples follow a Gaussian mixture distribution in a latent space. VPAC can not only be applied to datasets of discrete counts and normalized continuous data, but also scale up well to various data dimensionality, different dataset size and different data sparsity.
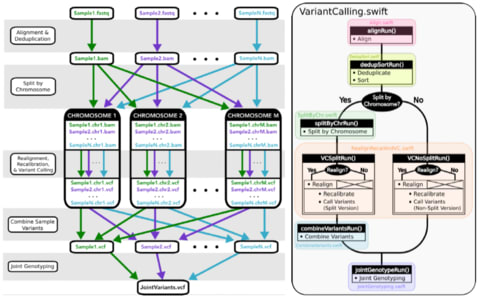
□ Managing genomic variant calling workflows with Swift/T:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/18/524645.full.pdf
a wrapper for the GATK-based variant calling workflow using the Swift/T parallel scripting language. The greatest purported advantages of Swift/T are its high portability and ability to scale up to extreme petascale computation levels.

□ On the Complexity of Exact Pattern Matching in Graphs: Binary Strings and Bounded Degree:
>> https://arxiv.org/pdf/1901.05264.pdf
a simple conditional lower bound that, for any constant ε > 0, an O(|E|1−ε m)-time or an O(|E|m1−ε)-time algorithm for exact pattern matching, with node labels and patterns from a binary alphabet, cannot be achieved unless the Strong Exponential Time Hypothesis (SETH) is false.

As Illumina Stock Price Declined, Waterstone Capital Management LP Upped Holding. The stock increased 0.87% or $2.7 during the last trading session, reaching $312.75. About 1.02M shares traded. Illumina, Inc.(NASDAQ:ILMN) has risen 50.49% since January 22, 2018 and is uptrending.

□ E2M: A Deep Learning Framework for Associating Combinatorial Methylation Patterns with Gene Expression:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/22/527044.full.pdf
E2M includes two 1D convolution and maxpooling layers that increase the number of filters and reduce the dimension of the feature space. the proposed E2M framework outperforms multiclass logistics regression and 3-layer fully connected network in prediction accuracy.

□ A discriminative learning approach to differential expression analysis for single-cell RNA-seq:
>> https://www.nature.com/articles/s41592-018-0303-9
This method detects changes in transcript dynamics and in overall gene abundance in large numbers of cells to determine differential expression. When applied to transcript compatibility counts obtained via pseudoalignment, this approach provides a quantification-free analysis of 3′ single-cell RNA-seq that can identify previously undetectable marker genes.

□ LAmbDA: Label Ambiguous Domain Adaption Dataset Integration Reduces Batch Effects and Improves Subtype Detection:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/24/522474.full.pdf
a species- and dataset-independent transfer learning framework (LAmbDA). LAmbDA Feedforward 1 Layer Neural Network was the only method to correctly predict ambiguous cellular subtype labels compared to CaSTLe and MetaNeighbor.

□ MultiXcan: Integrating predicted transcriptome from multiple tissues improves association detection
>> https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1007889
MultiXcan integrates evidence across multiple panels using multivariate regression, which naturally takes into account the correlation structure.
□ GFAKluge: A C++ library and command line utilities for the Graphical Fragment Assembly formats:
>> http://joss.theoj.org/papers/d731f6dfc6b77013caaccfd8333c684a
□ Space-Efficient Computation of the LCP Array from the Burrows-Wheeler Transform:
>> https://arxiv.org/pdf/1901.05226.pdf
The procedure at the core of this algorithms can be used to enumerate suffix tree intervals in succinct space from the BWT, which is of independent interest. An engineered implementation of the first algorithm on DNA alphabet induces the LCP of a large (16 GiB) collection of short (100 bases) reads at a rate of 2.92 megabases per second using in total 1.5 Bytes per base in RAM.
□ NORTH: a highly accurate and scalable Naive Bayes based ORTHologous gene clustering algorithm:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/23/528323.full.pdf
NORTH involves a novel combination of tools from natural language processing and machine learning with the biological basis of orthologs. the BLAST search based protocols deeply resemble a “text classification” problem. employing the robust bag-of-words model accompanied by a Naive Bayes classifier to cluster the orthologous genes.
□ MaXLinker: proteome-wide cross-link identifications with high specificity and sensitivity:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/23/526897.full.pdf
a novel “MS3-centric” approach for cross-link identification and implemented it as a search engine called MaXLinker. MaXLinker significantly outperforms XlinkX with up to 18-fold lower false positive rate, and the results in up to 31% more cross-links, demonstrating its superior sensitivity and specificity.
□ 2.7.0a release introduces STARsolo, a turnkey solution for analyzing droplet single-cell RNA sequencing (e.g. 10X) built directly into the STAR code.
It produces gene counts nearly identical to 10X CellRanger. At the same time, STARsolo is ~10 times faster than the CellRanger.
□ Karen Miga emphasizes the importance and attainability of the full assembly of one human genome with the technology available today.
>> #Genomics2020
□ A Single-Molecule Long-Read Survey of Human Transcriptomes using LoopSeq Synthetic Long Read Sequencing:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/28/532135.full.pdf
Reconstructed transcript sequences are characterized and annotated using SQANTI, an analysis pipeline for assessing the sequence quality of long-read transcriptomes. The results demonstrate that LoopSeq synthetic long-read sequencing can reconstruct contigs up to 3,900nt full-length transcripts using tissue extracted RNA, as well as identify novel splice variants of known junction donors and acceptors.
□ Block HSIC Lasso: model-free biomarker detection for ultra-high dimensional data:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/28/532192.full.pdf
block HSIC Lasso, a nonlinear feature selector. Block HSIC Lasso retains the properties of HSIC Lasso while extending its applicability to larger datasets. he memory complexity goes from O(dn2) down to O(dnB), where B << n is the block size. Moreover, as opposed to MapReduce HSIC Lasso, the optimization problem of the block HSIC Lasso remains convex.
<br />
□ VarSight: Prioritizing Clinically Reported Variants with Binary Classification Algorithms:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/28/532440.full.pdf
the application of classification algorithms that ingest variant predictions along with phenotype information for predicting whether a variant will ultimately be clinically reported and returned to a patient.
□ Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes
>> https://www.biorxiv.org/content/10.1101/530824v1
Comparison of Illumina+OxfordNanopore vs. Illumina+PacBio: "Both strategies facilitate high-quality genome reconstruction. Combining ONT and Illumina reads fully resolved most genomes without additional manual steps, and at a lower cost per isolate in our setting."
□ James Blachly
>> https://twitter.com/jamesblachly/status/1089172835992514564
HDF5 is a random access container file format (like a hierarchical filesystem within a file) used by both ONT for signal + basecalls, as well as e.g. @lpachter ‘s kallisto storing abundance estimates + bootstraps + metadata.
□ Quantum Genomics Société Anonyme (ENXTPA:ALQGC), Loral Space & Communications Inc. (NasdaqGS:LORL): Stock Earnings Analysis & Valuation Update:
>> https://steeleherald.com/2019/01/26/quantum-genomics-societe-anonyme-enxtpaalqgc-loral-space-communications-inc-nasdaqgslorl-stock-earnings-analysis-valuation-update/
The Earnings to Price yield of Quantum Genomics Société Anonyme (ENXTPA:ALQGC) is -0.163466. Quantum Genomics Société Anonyme has a Piotroski F-Score of 1.

□ MetaSeek: a data discovery and analysis tool for biological sequencing data:
>> https://github.com/MetaSeek-Sequencing-Data-Discovery/metaseek
MetaSeek scrapes metadata from the sequencing data repositories, cleaning and filling in missing or erroneous metadata, and stores the cleaned and structured metadata in the MetaSeek database.
□ ensembldb: an R package to create and use Ensembl-based annotation resources:
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz031/5301311
The ensembldb Bioconductor package retrieves and stores Ensembl-based genetic annotations and positional information, and furthermore offers identifier conversion and coordinate mappings for gene-associated data.
□ GIGI2: A Fast Approach for Parallel Genotype Imputation in Large Pedigrees:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/29/533687.full.pdf
GIGI2 performs imputation with computational time reduced by at least 25x on one thread and 120x on eight threads. The memory usage of GIGI2 is reduced by at least 30x. This reduction is achieved by implementing better memory layout and a better algorithm for solving the Identity by Descent graphs, as well as with additional features, including multithreading.
□ BUSseq: Flexible Experimental Designs for Valid Single-cell RNA-sequencing Experiments Allowing Batch Effects Correction:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/29/533372.full.pdf
BUSseq is an interpretable Bayesian hierarchical model that closely follows the data-generating mechanism of scRNA-seq experiments. BUSseq can simultaneously correct batch effects, cluster cell types, impute missing data caused by dropout events, and detect differentially expressed genes without requiring a preliminary normalization step.

□ RWBRMDA: Integrating random walk and binary regression to identify novel miRNA-disease association:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2640-9
a computational model of Random Walk and Binary Regression-based MiRNA-Disease Association prediction (RWBRMDA). RWBRMDA extracted features for each miRNA from random walk with restart on the integrated miRNA similarity network for binary logistic regression to predict potential miRNA-disease associations.

□ PRINCIPAL COMPONENT ANALYSIS AND ITS GENERALIZATIONS FOR ANY TYPE SEQUENCE (PCA-SEQ):
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/30/535112.full.pdf
For any sequence, including molecular, PCA-Seq without any additional assumptions allows to get its principal components in a numerical form and visualize them in the form of phase portraits. Long-term experience of SSA application for numerical data gives all reasons to believe that PCA-Seq will be not less useful in the analysis of non-numerical data, especially in hypothesizing.
□ Principal Stratification on a Latent Variable (fitting a multilevel model using Stan)
>> https://statmodeling.stat.columbia.edu/2019/01/31/principal-stratification-latent-variable-fitting-multilevel-model-using-stan/










※コメント投稿者のブログIDはブログ作成者のみに通知されます