(Generative Art by gen_ericai)
(Generative Art by gen_ericai)
□ scKINETICS: inference of regulatory velocity with single-cell transcriptomics data
>> https://academic.oup.com/bioinformatics/article/39/Supplement_1/i394/7210448
scKINETICS (Key regulatory Interaction NETwork for Inferring Cell Speed), an integrative algorithm which combines inference of regulatory network structure with robust de novo estimation of gene expression velocity under a model of causal, regulation-driven dynamics.
scKINETICS models changes in cellular phenotype with a joint system of dynamic equations governing the expression of each gene as dictated by these regulators within a genome-wide GRN.
scKINETICS uses an expectation-maximization approach derived to learn the impact of each regulator on its target genes, leveraging biologically-motivated priors from epigenetic data, gene-gene co-expression, and constraints on cells’ future states imposed by the phenotypic manifold.

□ scTranslator: A pre-trained large language model for translating single-cell transcriptome to proteome
>> https://www.biorxiv.org/content/10.1101/2023.07.04.547619v1
scTranslator, which is align-free and generates absent single-cell proteome by inferring from the transcriptome. scTranslator achieves a general knowledge of RNA-protein interactions by being pre-trained on substantial amounts of bulk and single-cell data.
By innovatively introducing the re-index Gene Positional Encoding (GPE) module into Transformer, scTranslator can infer any protein determined by the user's query, as the GPE module has comprehensive coverage of all gene IDs and reserves another 10,000 positions for new findings.
sTranslator does not employ an autoregressive decoder. The generative style decoder of sTranslator predicts the long sequences at one forward operation, thereby improving the inference efficiency of long-sequence predictions.

□ HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution
>> https://arxiv.org/abs/2306.15794
HyenaDNA, a genomic foundation model pretrained on the human reference genome with context lengths of up to 1 million tokens at the single nucleotide-level – an up to 500x increase over previous dense attention-based models.
HyenaDNA scales sub-quadratically in sequence length (training up to 160x faster than Transformer), uses single nucleotide tokens, and has full global context at each layer. For comparison they construct embeddings using DNABERT (5-mer) and Nucleotide Transformer.
In HyenaDNA block architecture, a Hyena operator is composed of long convolutions and element-wise gate layers. The long convolutions are parameterized implicitly via an MLP. The convolution is evaluated using a Fast Fourier Transform convolution with time complexity O(Llog2 L).

□ Co-linear Chaining on Pangenome Graphs
>> https://www.biorxiv.org/content/10.1101/2023.06.21.545871v1
PanAligner, an end-to-end sequence-to-graph aligner using seeding and alignment code from Minigraph. An iterative chaining algorithm which builds on top of the known algorithms for DAGs.
The dynamic programming-based chaining algorithms developed for DAGs exploit the topological ordering of vertices, but such an ordering is not available in cyclic graphs. Computing the width and a minimum path cover can be solved in polynomial time for DAGs but is NP-hard.
The walk corresponding to the optimal sequence-to-graph alignment can traverse a vertex multiple times if there are cycles. Accordingly, a chain of anchors should be allowed to loop through vertices.
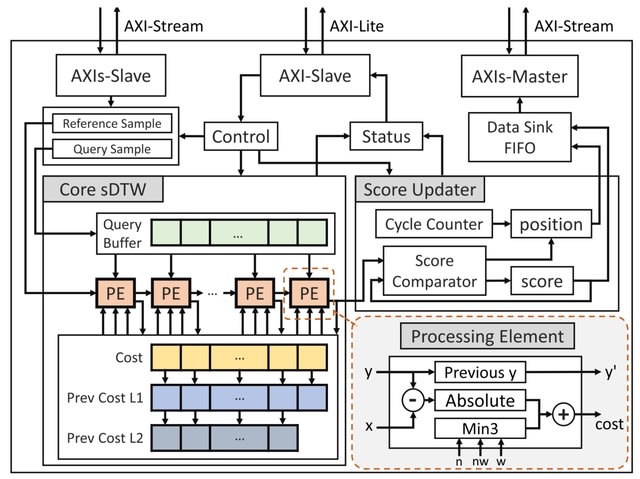
□ HARU: Efficient real-time selective genome sequencing on resource-constrained devices
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad046/7217084
HARU (Hardware Accelerated Read Until), a software-hardware codesign system for raw signal-alignment Read Until that uses the memory-efficient subsequence dynamic time warping (sDTW) hardware accelerator for high-throughput signal mapping.
HARU tackles the computational bottleneck by accelerating the sDTW algorithm with field-programmable gate arrays (FPGAs). HARU performs efficient multithreaded batch-processing for signal preparation in conjunction with the sDTW accelerator.


□ BioAlpha: BioTuring GPU-accelerated single-cell data analysis pipeline
>> https://alpha.bioturing.com/
BioTuring Alpha’s single-cell pipeline has reported an end-to-end runtime that was 169 times and 121 times faster than Scanpy and Seurat, respectively. BioAlpha enables reading a sparse matrix up to 150 times faster compared to scipy in Python and Matrix in R.
BioAlpha provides a highly optimized GPU implementation of NN-descent to unlock unprecedented performance. BioAlpha finishes this step 270 times faster than scanpy. Louvain Alpha achieves an impressive 2000x speed-up for some dataset while maintains similar clustering quality.

□ MOWGAN: Scalable Integration of Multiomic Single Cell Data Using Generative Adversarial Networks
>> https://www.biorxiv.org/content/10.1101/2023.06.26.546547v2
MOWGAN learns the structure of single assays and infers the optimal couplings between pairs of assays. MOWGAN generates synthetic multiomic datasets that can be used to transfer information among the measured assays by bridging.
A WGAN-GP is a generative adversarial network that uses the Wasserstein (or Earth-Mover) loss function and a gradient penalty to achieve Lipschitz continuity. MOWGAN's generator outputs a synthetic dataset where cell pairing is introduced across multiple modalities.
MOWGAN's inputs are molecular layers embedded into a feature space having the same dimensionality. To capture local topology within each dataset, cells in each embedding are sorted by the first component of its Laplacian Eigenmap.

□ PanGenome Research Tool Kit (PGR-TK): Multiscale analysis of pangenomes enables improved representation of genomic diversity for repetitive and clinically relevant genes
>> https://www.nature.com/articles/s41592-023-01914-y
PGR-TK provides pangenome assembly management, query and Minimizer Anchored Pangenome (MAP) Graph Generation. Several algorithms and data structures used for the Peregrine Genome Assembler are useful for Pangenomics analysis.
PGR-TK uses minimizer anchors to generate pangenome graphs at different scales without more computationally intensive sequence-to-sequence alignment. PGR-TK decomposes tangled pangenome graphs, and can easily project the linear genomics sequence onto the principal bundles.

□ Velvet: Deep dynamical modelling of developmental trajectories with temporal transcriptomics
>> https://www.biorxiv.org/content/10.1101/2023.07.06.547989v1
velvet, a deep learning framework that extends beyond instantaneous velocity estimation by modelling gene expression dynamics through a neural stochastic differential equation system within a variational autoencoder.
Velvet trajectory distributions capture dynamical aspects such as decision boundaries between alternative fates and correlative gene regulatory structure.
velvetSDE, that infers global dynamics by embedding the learnt vector field in a neural stochastic differential equation (nSDE) system that is trained to produce accurate trajectories that stay within the data distribution.
velvetSDE's predicted trajectory distributions map the commitment of cells to specific fates over time, and can faithfully conserve known trends while capturing correlative structures between related genes that are not observed in unrelated genes.

□ HEAL: Hierarchical Graph Transformer with Contrastive Learning for Protein Function Prediction
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btad410/7208864
HEAL utilizes graph contrastive learning as a regularization technique to maximize the similarity between different views of the graph representation. HEAL is capable of finding functional sites through class activation mapping.
HEAL captures structural semantics using a hierarchical graph Transformer, which introduces a range of super-nodes mimicing functional motifs to interact with nodes. These semantic-aware super-node embeddings are aggregated w/ varying emphasis to produce a graph representation.
<brr />

□ GRADE-IF: Graph Denoising Diffusion for Inverse Protein Folding
>> https://arxiv.org/abs/2306.16819
GRADE-IF, a diffusion model backed by roto-translation equivariant graph neural network for inverse folding. It stands out from its counterparts for its ability to produce a wide array of diverse sequence candidates.
As a departure from conventional uniform noise in discrete diffusion models, GRADE-IF encodes the prior knowledge of the response of As to evolutionary pressures by the utilization of Blocks Substitution Matrix as the translation kernel.

□ Grid Codes versus Multi-Scale, Multi-Field Place Codes for Space
>> https://www.biorxiv.org/content/10.1101/2023.06.18.545252v1
An evolutionary optimization of several multi-scale, multi-field place cell networks and compare the results against a single-scale, single-field as well as against a simple grid code.
A new dynamic MSMF model (D-MSMF) composed of a dynamic number of attractor networks. The model has the general architecture of a CAN but dos not fully comply with all properties of either a continuous or a discrete attractor network, settling it somewhere in between.

□ scTour: a deep learning architecture for robust inference and accurate prediction of cellular dynamics
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02988-9
scTour provides two main functionalities in deciphering cellular dynamics in a batch-insensitive manner: inference and prediction. For inference, the time neural network in scTour allows estimates of cell-level pseudotime along the trajectory.
scTour leverages a neural network to assign a time point to each cell in parallel to the neural network for latent variable parameterization. The learned differential equation by another neural network provides an alternative way of inferring the transcriptomic vector field.

□ Protein Discovery with Discrete Walk-Jump Sampling
>> https://arxiv.org/abs/2306.12360
Resolving difficulties in training and sampling from a discrete generative model by learning a smoothed energy function, sampling from the smoothed data manifold with Langevin Markov chain Monte Carlo (MCMC), and projecting back to the true data manifold with one-step denoising.
The Discrete Walk-Jump Sampling formalism combines the maximum likelihood training of an energy-based model and improved sample quality of a score-based model. This method outperforms autoregressive large language models, diffusion, and score-based baselines.

□ Multi pathways temporal distance unravels the hidden geometry of network-driven processes
>> https://www.nature.com/articles/s42005-023-01204-1
A multi-pathways temporal distance between nodes that overcomes the limitation of focussing only on the shortest path. This metric predicts the latent geometry induced by the dynamics in which the signal propagation resembles the traveling wave solution of reaction-diffusion systems.
This framework naturally encodes the concerted behavior of the ensemble of paths connecting two nodes in conveying perturbations. Embedding targets nodes in the vector space induced by this metric reveals the intuitive, hidden geometry of perturbation propagation.

□ Clustering the Planet: An Exascale Approach to Determining Global Climatype Zones
>> https://www.biorxiv.org/content/10.1101/2023.06.27.546742v1
Using a GPU implementation of the DUO Similarity Metric on the Summit supercomputer, we calculated the pairwise environmental similarity of 156,384,190 vectors of 414,640 encoded elements derived from 71 environmental variables over a 50-year time span at 1km2 resolution.
GPU matrix-matrix (GEMM) kernels were optimized for the GPU architecture and their outputs were managed through aggressive concurrent MPI rank CPU communication, calculations, and transfers.
Using vector transformation and highly optimized operations of generalized distributed dense linear algebra, calculation of all-vector-pairs similarity resulted in 5.07 x 1021 element comparisons and reached a peak performance of 2.31 exaflops.

□ Phantom oscillations in principal component analysis
>> https://www.biorxiv.org/content/10.1101/2023.06.20.545619v1
The “phantom oscillations” are a statistical phenomenon that explains a large fraction of variance despite having little to no relationship with the underlying data.
In one dimension, such as timeseries, phantom oscillations resemble sine waves or localized wavelets, which become Lissajous-like neural trajectories when plotted against each other.
In multiple dimensions, they resemble modes of vibration like a stationary or propagating wave, dependent on the spatial geometry of how they are sampled. Phantom oscillations may also occur on any continuum, such as a graph or a manifold in high-dimensional space.

□ InGene: Finding influential genes from embeddings of nonlinear dimension reduction techniques
>> https://www.biorxiv.org/content/10.1101/2023.06.19.545592v1
While non-linear dimensionality reduction techniques such as tSNE and UMAP are effective at visualizing cellular sub-populations in low-dimensional space, they do not identify the specific genes that influence the transformation.
InGene, in principle, can be applied to any linear or nonlinear dimension reduction method to extract relevant genes. InGene poses the whole problem of cell type-specific gene finding as a single bi-class classification problem.

□ Cofea: correlation-based feature selection for single-cell chromatin accessibility data
>> https://www.biorxiv.org/content/10.1101/2023.06.18.545397v1
Cofea, a correlation-based framework to select biologically informative features of scCAS data via placing emphasis on the correlation among features. Cofea obtains a peak-by-peak correlation matrix after a stepwise preprocessing
approach.
Cofea establishes a fitting relationship between the mean and mean square values of correlation coefficients to reveal a prevailing pattern observed across the majority of features, and selects features that deviate from the established pattern.

□ Stochastic Collapse: How Gradient Noise Attracts SGD Dynamics Towards Simpler Subnetworks
>> https://arxiv.org/abs/2306.04251
Revealing a strong implicit bias of stochastic gradient descent (SGD) that drives overly expressive networks to much simpler subnetworks, thereby dramatically reducing the number of independent parameters, and improving generalization.
SGD exhibits a property of stochastic attractivity towards these simpler invariant sets. A sufficient condition for stochastic attractivity based on a competition between the loss landscape's curvature around the invariant set and the noise introduced by stochastic gradients.
An increased level of noise strengthens attractivity, leading to the emergence of attractive invariant sets associated with saddle-points or local maxima of the train loss.
Empirically, the existence of attractive invariant sets in trained deep neural networks, implying that SGD dynamics often collapses to simple subnetworks with either vanishing or redundant neurons.

□ JTK: targeted diploid genome assembler
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad398/7206882
JTK, a megabase-scale diploid genome assembler. It first randomly samples kilobase-scale sequences (called “chunks”) from the long reads, phases variants found on them, and produces two haplotypes.
JTK utilizes chunks to capture SNVs and SVs simultaneously. JTK finds SNVs on these chunks and separates the chunks into each copy. JTK introduces each possible SNV to the chunk and accepts it as an actual SNV if the alignment scores of many reads increase.
JTK determines the order of these separated copies in the target region. Then, it produces the assembly by traversing the graph. JTK constructs a partially phased assembly graph and resolves the remaining regions to get a fully phased assembly.

□ Deep Language Networks: Joint Prompt Training of Stacked LLMs using Variational Inference
>> https://arxiv.org/abs/2306.12509
LLMs as language layers in a Deep Language Network (DLN). The learnable parameters of each layer are the associated natural language prompts and the LLM at a given layer receives as input the output of the LLM at the previous layer, like in a traditional deep network.
DLN-2 provides a boost to DLN-1. On Nav., DLN-2 successfully outperforms the GPT-4 0-shot baseline and GPT-4 ICL by 5% accuracy. On Date., DLN-2 further improves the performance of DLN-1, outperforming all single layer networks, but is far from matching GPT-4, even in O-shot.

□ ExplaiNN: interpretable and transparent neural networks for genomics
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02985-y
ExplaiNN, a fully interpretable and transparent deep learning model for genomic tasks inspired by NAMs. ExplaiNN computes a linear combination of multiple independent CNNs, each consisting of one convolutional layer with a single filter followed by exponential activation.
ExplaiNN provides local interpretability by multiplying the output of each unit by the weight of that unit for each input sequence. Architecturally, ExplaiNN models are constrained to only capturing homotypic cooperativity, excl. heterotypic interactions between pairs of motifs.

□ Read2Tree: Inference of phylogenetic trees directly from raw sequencing reads
>> https://www.nature.com/articles/s41587-023-01753-4
Read2Tree directly processes raw sequencing reads into groups of corresponding genes and bypasses traditional steps in phylogeny inference, such as genome assembly, annotation and all-versus-all sequence comparisons, while retaining accuracy.
Read2Tree can process the input genomes in parallel, and scales linearly with respect to the number of input genomes. Read2Tree is 10–100 times faster than assembly-based approaches—the exception being when sequencing coverage is high and reference species very distant.

□ S-leaping: an efficient downsampling method for large high-throughput sequencing data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad399/7206878
S-leaping, a method that focuses on downsampling of large datasets by approximating reservoir sampling. By applying the concept of leaping to downsampling, s-leaping simplifies the sampling procedure and reduces the average number of random numbers it requires.
S-leaping is a hybrid method that combines Algorithm R and an efficient approximate next selection method. It follows Algorithm R for the first 2 k-th elements when the probability of selecting each element is at least 0.5.

□ ISRES+: An improved evolutionary strategy for function minimization to estimate the free parameters of systems biology models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad403/7206879
ISRES+, an upgraded algorithm that builds on the Improved Evolutionary Strategy by Stochastic Ranking (ISRES). ISRES+ employs two gradient-based strategies: Linstep and Newton step, to understand the features of the fitness landscape by sharing information between individuals.
The Linstep is a first-order linear least squares fit method which generates offspring by approximating the structure of the fitness landscape by fitting a hyperplane. Linstep could potentially overshoot a minimum basin in a phenomenon known as gradient hemistitching.
The Newton step is a second-order linear least squares fit method which generates new offspring by approximating the structure of the fitness landscape around the O(n2) individuals around the fittest individual in every generation by a quadric hypersurface.

□ Pangene: Constructing a pangenome gene graph
>> https://github.com/lh3/pangene
Pangene is a command-line tool to construct a pangenome gene graph. In this graph, a node repsents a marker gene and an edge between two genes indicates their genomic adjaceny on input genomes.
Pangene takes the miniprot alignment between a protein set and multiple genomes and produces a graph in the GFA format. It attempts to reduce the redundancy in the input proteins and filter spurious alignments while preserving close but non-identical paralogs.