








 (By @muratpak)
(By @muratpak)己が辿った経路は遥か遠景を辿るように、その果てを常に朝霞の向こうに溶かしている。
だが、この画を近傍から映し撮ることが叶う者たちは、
滲んだ道の先に未だ捉えぬ輪郭を描き出すことが出来る。

□ MultiVERSE: a multiplex and multiplex-heterogeneous network embedding approach
>> https://www.nature.com/articles/s41598-021-87987-1
MultiVERSE, an extension of the VERSE framework using Random Walks with Restart on Multiplex (RWR-M) and Multiplex-Heterogeneous (RWR-MH) networks. MultiVERSE is a fast and scalable method to learn node embeddings from multiplex and multiplex-heterogeneous networks.
Spherical K-means clustering is well-adapted to high-dimensional clustering. MultiVERSE effectively captures node properties and a better representation of the topological structure of the multiplex network as RWR-M applies a random walk in pseudo-infinite time.

□ scShaper: ensemble method for fast and accurate linear trajectory inference from single-cell RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2021.05.03.442435v1.full.pdf
scShaper is able to infer accurate trajectories for a variety of nonlinear mathematical trajectories, including many for which the commonly used principal curves method fails.
scShaper smooths the ensemble pseudotime using local regression (LOESS). The clustering is performed using the k-means algorithm, and the result is permuted using a special case of Kruskal's algorithm.
scShaper is based on graph theory and solves the shortest Hamiltonian path of a clustering, utilizing a greedy algorithm to permute clusterings computed using the k-means method to obtain a set of discrete pseudotimes.

□ PseudotimeDE: inference of differential gene expression along cell pseudotime with well-calibrated p-values from single-cell RNA sequencing data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02341-y
PseudotimeDE uses subsampling to estimate pseudotime inference uncertainty and propagates the uncertainty to its statistical test for DE gene identification.
PseudotimeDE fits NB-GAM or zero-inflated negative binomial GAM to every gene in the dataset to obtain a test statistic that indicates the effect size of the inferred pseudotime on the GE. Pseudotime fits a Gamma distribution or a mixture of two Gamma distributions.

□ QuASeR: Quantum Accelerated de novo DNA sequence reconstruction
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0249850
QuASeR, a reference-free DNA sequence reconstruction implementation via de novo assembly on both gate-based and quantum annealing platforms.
Each one of the four steps of the implementation (TSP, QUBO, Hamiltonians and QAOA) is explained with a proof-of-concept example to target both the genomics research community and quantum application developers in a self-contained manner.
This is the target algorithm for which the quantum kernel is formulated. The implementation and results on executing the algorithm from a set of DNA reads to a reconstructed sequence, on a gate-based quantum simulator, the D-Wave quantum annealing simulator.

□ XENet: Using a new graph convolution to accelerate the timeline for protein design on quantum computers
>> https://www.biorxiv.org/content/10.1101/2021.05.05.442729v1.full.pdf
XENet is a message-passing GNN that simultaneously accounts for both the incoming and outgoing neighbors of each node, such that a node’s representation is based on the messages it receives as well as those it sends.
XENet is the attempt to engineer a new GNN layer that makes further use of the edge tensors, including updating their features as the result of the convolution.
XENet's goal was to find the set of rotamers that minimizes the proteincomputed energy, measured in Rosetta Energy Units (REU). Rosetta does this using simulated annealing in a process.

□ SCALEX: Construction of continuously expandable single-cell atlases through integration of heterogeneous datasets in a generalized cell-embedding space
>> https://www.biorxiv.org/content/10.1101/2021.04.06.438536v1.full.pdf
SCALEX (Single-Cell ATAC-seq Analysis via Latent feature Extraction) disentangles batch-related components away from batch-invariant components of single-cell data.
SCALEX implements a batch-free encoder and a batch-specific decoder in an asymmetric VAE framework. SCALEX renders the encoder to function as a data projector that projects single cells of different batches into a generalized, batch-invariant cell-embedding space.

□ Recursive MAGUS: scalable and accurate multiple sequence alignment
>> https://www.biorxiv.org/content/10.1101/2021.04.09.439137v1.full.pdf
MAGUS uses the GCM (Graph Clustering Merger) technique to combine an arbitrary number of subalignments, which allows MAGUS to align large numbers of sequences with highly competitive accuracy and speed.
Recursive MAGUS allowing it to scale from 50,000 to a full million sequences. Instead of automatically aligning our subsets with MAFFT, subsets larger than a threshold are recursively aligned with MAGUS.
Recursive MAGUS generates the guide tree with Clustal Omega’s initial tree method, MAFFT’s PartTree initial tree method, and FastTree’s minimum evolution tree. In extremis, the dataset can be decomposed randomly for maximum speed.
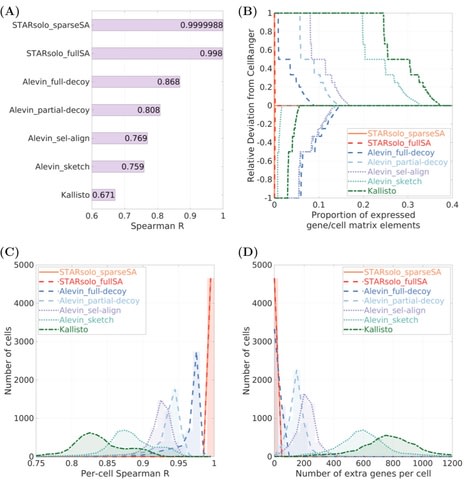
□ STARsolo: accurate, fast and versatile mapping/quantification of single-cell and single-nucleus RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2021.05.05.442755v1.full.pdf
STARsolo is built directly into the RNA-seq aligner STAR, and can be run similarly to standard STAR bulk RNA-seq alignment, specifying additionally the single-cell parameters such as barcode geometry and passlist.
In STARsolo, read mapping, read-to-gene assignment, cell barcode demultiplexing and UMI collapsing are tightly integrated, avoiding input/output bottlenecks and boosting the processing speed.

□ DAVAE: Efficient and scalable integration of single-cell data using domain-adversarial and variational approximation
>> https://www.biorxiv.org/content/10.1101/2021.04.06.438733v1.full.pdf
Domain-Adversarial and Variational Auto-Encoder (DAVAE), to fit the normalized gene expression into a non-linear model, which transforms a latent variable z into the expression space with a non-linear function, a KL regularizier and a domain-adversarial regularizier.
The Gradient Reversal Layer enables the adversarial mechanism, which takes the gradient from the subsequence and changes its sign before passing it to the preceding layer. The latent variables in the lower dimensional space can be used for trajectory inference across modalities.

□ scDART: Learning latent embedding of multi-modal single cell data and cross-modality relationship simultaneously
>> https://www.biorxiv.org/content/10.1101/2021.04.16.440230v1.full.pdf
scDART (single cell Deep learning model for ATAC-Seq and RNA-Seq Trajectory integration) is a scalable deep learning framework that embed the two data modalities, scRNA-seq and scATAC-seq data, into a shared low-dimensional latent space while preserving cell trajectory structures.
scDART learns a nonlinear function represented by a neural network encoding the cross-modality relationship simultaneously when learning the latent space representations of the integrated dataset.
scDART’s gene activity function module is a fully-connected NN. It encodes the nonlinear regulatory relationship b/n regions / genes. the projection module takes in the scRNA-seq count matrix and the pseudo- scRNA-seq matrix, and generates the latent embedding of both modalities.

□ stPlus: a reference-based method for the accurate enhancement of spatial transcriptomics
>> https://www.biorxiv.org/content/10.1101/2021.04.16.440115v1.full.pdf
stPlus is robust and scalable to datasets of diverse gene detection sensitivity levels, sample sizes, and number of spatially measured genes.
stPlus first augments spatial transcriptomic data and combines it with reference scRNA-seq data. The data is then jointly embedded using an auto-encoder. Finally, stPlus predicts the expression of spatially unmeasured genes based on weighted k-NN.

□ SENSV: Detecting Structural Variations with Precise Breakpoints using Low-Depth WGS Data from a Single Oxford Nanopore MinION Flowcell
>> https://www.biorxiv.org/content/10.1101/2021.04.20.440583v1.full.pdf
SENSV, by integrating several efficient algorithmic techniques, including SV-aware alignment (SV-DP), analysis of sequencing depth information, and sophisticated verification via re-alignment.
SENSV can effectively utilize 4x ONT whole genome sequencing data to detect heterozygous structural variations with superior sensitivity, precision and breakpoint resolution.

□ Simplitigs as an efficient and scalable representation of de Bruijn graphs
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02297-z
Simplitigs correspond to vertex-disjoint paths covering the graph but relax the unitigs’ restriction of stopping at branching nodes.
an algorithm for rapid simplitig computation from a k-mer set and implement it in a tool called ProphAsm, which proceeds by loading a k-mer set into memory and a greedy enumeration of maximal vertex-disjoint paths in the associated de Bruijn graph.

□ TReNCo: Topologically associating domain (TAD) aware regulatory network construction
>> https://www.biorxiv.org/content/10.1101/2021.04.27.441672v1.full.pdf
TReNCo, a memory-lean method utilizing epigenetic marks of enhancer and promoter activity, and gene expression to create context-specific transcription factor-gene regulatory networks.
TReNCo utilizes TAD boundaries as a hard cutoff, instead of distance based, to efficiently create context-specific TF-gene regulatory networks, and utilize dynamic programming to factor matrices within TADs and combine network into a full adjacency matrix for a regulatory graph.
□ PANDORA-seq expands the repertoire of regulatory small RNAs by overcoming RNA modifications
>> https://www.nature.com/articles/s41556-021-00652-7
PANDORA-seq (panoramic RNA display by overcoming RNA modification aborted sequencing), employing a combinatorial enzymatic treatment to remove key RNA modifications that block adapter ligation and reverse transcription.
PANDORA-seq identified abundant modified sncRNAs—transfer RNA (tsRNAs) and ribosomal RNA-derived small RNAs (rsRNAs). tsRNAs and rsRNAs that are downregulated during somatic cell reprogramming impact cellular translation in ESCs, suggesting a role in lineage differentiation.

□ Modular, efficient and constant-memory single-cell RNA-seq preprocessing
>> https://www.nature.com/articles/s41587-021-00870-2
a single experiment can look at 100,000 cells and measure information from hundreds of thousands of transcripts (fragments of RNA produced when a gene is active), resulting in tens of billions of sequenced fragments.
The workflow is based on the kallisto and bustools programs, and is near optimal in speed with a constant memory requirement providing scalability for arbitrarily large datasets.

□ Effect of imputation on gene network reconstruction from single-cell RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2021.04.13.439623v1.full.pdf
an inflation of gene-gene correlations that affects the predicted network structures and may decrease the performance of network reconstruction in general.Evaluating the combination between imputation and network inference on different datasets results in a cubic matrix.
Cubic evaluation matrix consists of seven cell types from experimental scRNAseq data, four imputation methods and three network reconstruction algorithms using the BEELINE framework.

□ RCSL: Clustering single-cell RNA-seq data by rank constrained similarity learning
>> https://www.biorxiv.org/content/10.1101/2021.04.12.439254v1.full.pdf
RCSL considers both local similarity and global similarity among the cells to discern the subtle differences among cells of the same type as well as larger differences among cells of different types.
RCSL uses Spearman’s rank correlations of a cell’s expression vector with those of other cells to measure its global similarity, and adaptively learns neighbour representation of a cell as its local similarity.
RCSL automatically estimates the number of cell types defined in the similarity matrix, and identifies them by constructing a block-diagonal matrix, such that its distance to the similarity matrix is minimized.
□ UCell: robust and scalable single-cell gene signature scoring
>> https://www.biorxiv.org/content/10.1101/2021.04.13.439670v1.full.pdf
UCell scores, based on the Mann-Whitney U statistic, are robust to dataset size and heterogeneity, and their calculation demands relatively less computing time and memory than other available methods, enabling the processing of large datasets (10^5 cells).
UCell scores depend only on the relative gene expression in individual cells and are therefore not affected by dataset composition. UCell can be applied to any cell vs. gene data matrix, and includes functions to directly interact with Seurat objects.
□ AFLAP: assembly-free linkage analysis pipeline using k-mers from genome sequencing data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02326-x
AFLAP generates ultra-dense genetic maps based on single-copy k-mers without reference to a genome assembly. This approach to linkage analysis does not require reads to be mapped and variants called against a reference assembly for marker identification.
Assembly-free linkage analysis pipeline (AFLAP) enables the construction of accurate genotype tables resulting in high-quality genetic maps for any organism using a segregating population sequenced to adequate depth.
□ Cooperative Sequence Clustering and Decoding for DNA Storage System with Fountain Codes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab246/6255306
DNA Fountain, a strategy for DNA storage devices that approaches the Shannon capacity while providing strong robustness against data corruption. The strategy harnesses fountain codes which allows reliable unicasting of information over channels that are subject to dropouts.
the decoding process focusing on the cooperation of key components: Hamming-distance based clustering, discarding of abnormal sequence reads, Reed-Solomon (RS) error correction as well as detection, and quality score-based ordering of sequences.

□ Dynamic model updating (DMU) approach for statistical learning model building with missing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04138-z
DMU approach divides the dataset with missing values into smaller subsets of complete data followed by preparing and updating the Bayesian model from each of the smaller subsets.
DMU provides a different perspective of building models with missing data using available data as compared to the existing perspective in the literature of either removing missing data or imputing missing data. DMU does not depend on the association among the predictors.

□ LSH-GAN: Generating realistic cell samples for gene selection in scRNA-seq data: A novel generative framework
>> https://www.biorxiv.org/content/10.1101/2021.04.29.441920v1.full.pdf
a subsample of original data based on locality sensitive hashing (LSH) technique and augment this with noise distribution, which is given as input to the generator.
LSH-GAN can able to generate realistic samples in a faster way than the traditional GAN. This makes LSH-GAN more feasible to use in the feature (gene) selection problem of scRNA-seq data.

□ ScHiC-Rep: A novel framework for single-cell Hi-C clustering based on graph-convolution-based imputation and two-phase-based feature extraction
>> https://www.biorxiv.org/content/10.1101/2021.04.30.442215v1.full.pdf
ScHiC-Rep mainly contains two parts: data imputation and feature extraction. In the imputation part, a novel imputation workflow is proposed, including graph convolution-based, random walk with restart-based and genomic neighbor-based imputation.
A two-phase feature extraction method is proposed for learning the feature representation of a cell based on imputed single-cell Hi-C contact matrix, including linear phase for chromosome level and non-linear phase for cell level feature extraction.
□ q-mer analysis: a generalized method for analyzing RNA-Seq data.
>> https://www.biorxiv.org/content/10.1101/2021.05.01.424421v1.full.pdf
The q-mer analysis summarizes the RNA-Seq data using the "q-mer vector": the ratio of 4q kinds of q-length oligomer in the alignment data. by increasing the q value, q-mer analysis can produce the vector with a higher dimension than the one from the count-based method.
This "dimensionality increment" is the key point to describe the sample conditions more accurately than the count-based method does.
□ MDEC: Toward Multidiversified Ensemble Clustering of High-Dimensional Data: From Subspaces to Metrics and Beyond
>> https://ieeexplore.ieee.org/document/9426579/
a large number of diversified metrics by randomizing a scaled exponential similarity kernel, which are then coupled with random subspaces to form a large set of metric-subspace pairs.
Based on the similarity matrices derived from these metric-subspace pairs, an ensemble of diversified base clusterings can thereby be constructed.
an entropy-based criterion is utilized to explore the cluster-wise diversity in ensembles, Finally, based on diversified metrics, random subspaces, and weighted clusters, 3 specific ensemble clustering algorithms are presented by incorporating three types of consensus functions.

□ Chord: Identifying Doublets in Single-Cell RNA Sequencing Data by an Ensemble Machine Learning Algorithm
>> https://www.biorxiv.org/content/10.1101/2021.05.07.442884v1.full.pdf
Chord uses the AdBoost algorithm to integrate different methods for stable and accurate doublets filtered results.
Chord added a step, ‘overkill’, which first used different methods to evaluate the data, filtered out cells identified by any method, then simulated doublets by the remaining cells.
Chord’s input format is comma-separated expression matrix is a background-filtered, UMI-based matrix of a single sample. Chord will pre-process it according to the Seurat analysis pipeline. Chord can also directly accept object files generated by the Seurat analysis pipeline.
□ TieBrush: an efficient method for aggregating and summarizing mapped reads across large datasets
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab342/6272575
TieBrush, a software package designed to process very large sequencing datasets into a form that enables quick visual and computational inspection.
TieBrush can also be used as a method for aggregating data for downstream computational analysis, and is compatible with most software tools that take aligned reads as input.
□ Cellsnp-lite: an efficient tool for genotyping single cells
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btab358/6272512
cellsnp-lite was initially designed to pileup the expressed alleles in single-cell or bulk RNA-seq data, which can be directly used for donor deconvolution in multiplexed scRNA-seq data, which assigns cells to donors and detects doublets, even without genotyping reference.
Cellsnp-lite also provides a simplified user interface and better convenience that supports parallel computing, cell barcode and UMI tags.
cellsnp-lite does not aim to address the technical issues caused by sequencing platforms, e.g., uneven amplification in scDNA-seq and low coverage in scRNA-seq, but rather leaves them to downstream statistical modelling.
□ AMBARTI: Bayesian Additive Regression Trees for Genotype by Environment Interaction Models
>> https://www.biorxiv.org/content/10.1101/2021.05.07.442731v1.full.pdf
Additive Main Effects Bayesian Additive Regression Trees Interaction (AMBARTI) is a fully Bayesian semi-parametric machine learning approach that estimates main effects of genotypes and environments and interactions with an adapted regression tree-like structure.
AMBARTI allows the possibility of reasoning other than the ones obtained by models which consider the genotypic and environmental effects as linear and the interaction GxE in the maximum as bilinear.

□ Acorde: unraveling functionally-interpretable networks of isoform co-usage from single cell data
>> https://www.biorxiv.org/content/10.1101/2021.05.07.441841v1.full.pdf
acorde, an end-to-end pipeline to generate isoform co-expression networks and detect genes with co-Differential Isoform Usage (coDIU), and apply it to the study of isoform co-expression among seven neural broad cell types.
acorde successfully leveraged single-cell data by implementing percentile correlations, a metric designed to overcome single-cell noise and sparsity and provide high-confidence estimates of isoform-to-isoform correlation.
□ BiSulfite Bolt: A bisulfite sequencing analysis platform
>> https://academic.oup.com/gigascience/article/10/5/giab033/6272610
BSBolt incorporates bisulfite alignment logic directly within a forked version of BWA-MEM. BSBolt is designed around a single Burrows-Wheeler Transform (BWT) FM-index constructed from both bisulfite converted reference strands.
BSBolt includes a rapid and multi-threaded methylation caller, which outputs methylation calls in CGmap or bedGraph format implemented by BSSeeker2 and Bismark.
BSBolt was the fastest alignment tool across all simulation conditions, aligning close to 2.29 million reads per minute on average.
To facilitate end-to-end processing of bisulfite-sequencing data BSBolt includes utilities for read simulation utility and aggregation of methylation call files into a consensus matrix.
□ Prowler: A novel trimming algorithm for Oxford Nanopore sequence data
>> https://www.biorxiv.org/content/10.1101/2021.05.09.443332v1.full.pdf
Prowler (PROgressive multi-Window Long Read trimmer) was developed to remove low average Q-Score segments. The Prowler algorithm (Figure 1A) considers the quality distribution of the read by breaking the sequence into multiple non-overlapping windows.
Prowler out-performs Nanofilt as a QC program for ONT reads. The specific settings that are applied need to be considered when selecting trimming settings for Prowler due to the tradeoff between continuality and error rate of assemblies.

□ MAT2: Manifold alignment of single-cell transcriptomes with cell triplets
>> https://doi.org/10.1093/bioinformatics/btab260
MAT2 that aligns cells in the manifold space with a deep neural network employing contrastive learning strategy. with cell triplets defined based on known cell type annotations, the consensus manifold yielded by the alignment procedure is more robust.
by reconstructing both consensus and batch-specific matrices from the latent manifold space, MAT2 can be used to recover the batch- effect-free gene expression that can be used for downstream analysis.
□ NeuralPolish: a novel Nanopore polishing method based on alignment matrix construction and orthogonal Bi-GRU Networks
>> https://doi.org/10.1093/bioinformatics/btab354
a bi-directional GRU network is used to extract the sequence information inside each read by processing the alignment matrix row by row. the feature matrix is processed by another bi-directional GRU network column by column to calculate the probability distribution.
Finally, a CTC decoder generates a polished sequence with a greedy algorithm. NeuralPolish solves a large number of deletion errors at the cost of introducing some insertion errors, thereby reducing the overall error rate of the draft assembly.










※コメント投稿者のブログIDはブログ作成者のみに通知されます