









- [x] 私は常に、複雑な事象を他者に理解させたい時、可能な限り複雑さを損なわぬまま伝えられるよう試みている。

□ The Ramanujan Machine: Automatically Generated Conjectures on Fundamental Constants
>> https://arxiv.org/abs/1907.00205v4
The Ramanujan Machine, a novel and systematic approach that leverages algorithms for deriving mathematical formulas for fundamental constants and help reveal their underlying structure.
This algorithms find dozens of well-known as well as previously unknown continued fraction representations of π, e, Catalan's constant, and values of the Riemann zeta function.
The Ramanujan Machine using two algorithms that proved useful in finding conjectures: a Meet-In-The-Middle (MITM) algorithm and a Gradient Descent (GD) tailored to the recurrent structure of continued fractions.

□ LuxUS: DNA methylation analysis using generalized linear mixed model with spatial correlation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa539/5850399
LuxGLM Using Spatial correlation (LuxUS) based on generalized linear mixed model with spatial correlation structure. Savage-Dickey Bayes factor estimates are used for statistical testing of a covariate of interest.
LuxUS can model both binary and continuous covariates, and mixed model formulation enables including replicate and cytosine random effects. Spatial correlation is included to the model through a cytosine random effect correlation structure.
□ Meta-Align: A Novel HMM-based Algorithm for Pairwise Alignment of Error-Prone Sequencing Reads
>> https://www.biorxiv.org/content/10.1101/2020.05.11.087676v1.full.pdf
Meta-Align, a novel hidden Markov model (HMM)-based pairwise alignment algorithm, that aligns DNA sequences in the protein space, incorporating quality scores from the DNA sequences and allowing frameshifts caused by insertions and deletions.
A Viterbi algorithm over Meta-Align produces the optimal alignment of a pair of metagenomic reads taking into account all possible translating frames and gap penalties in both the protein space and the DNA space.
Meta-Align outperforms TBLASTX which compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database using the BLAST algorithm.
□ Sparsely-Connected Autoencoder (SCA) for single cell RNAseq data mining
>> https://www.biorxiv.org/content/10.1101/2020.05.26.117705v1.full.pdf
Sparsely-connected autoencoder (SCA) uses a single-layer autoencoder with sparse connections (representing known biological relationships) in order to attain a value for each gene set. SCA provides great flexibility for modelling biological phenomena.
Cell Stability Score is used to evaluate both SCA coherence. the effect of SCA input count table normalization on SCA encoding can be estimated using QCF and QCM scores. Thus, allowing to define the optimal condition to retrieve biological knowledge from the SCA encoded space.

□ Predicting Alignment Distances via Continuous Sequence Matching
>> https://www.biorxiv.org/content/10.1101/2020.05.24.113852v1.full.pdf
The CSM function is a modified local-global alignment algorithm using dynamic programming. a new embedding function specifically designed for biological sequences to map sequences into embedding vectors.
Continuous sequence matching (CSM), that embed variable length sequences in a continuous high-dimension embedding space using a list of short learned kernel sequences of same dimension.

□ stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues
>> https://www.biorxiv.org/content/10.1101/2020.05.31.125658v1.full.pdf
stLearn computes a distance measure using morphological similarity and neighbourhood smoothing.
stLearn uses a method to calculate transcriptional states by pseudo-space-time (PST) distance. PST distance is a function of physical distance (spatial distance) and gene expression distance (pseudotime distance) to estimate the pairwise similarity.

□ den-SNE/densMAP: Density-Preserving Data Visualization Unveils Dynamic Patterns of Single-Cell Transcriptomic Variability
>> https://www.biorxiv.org/content/10.1101/2020.05.12.077776v1.full.pdf
a general, differentiable measure of local density, called the “local radius”, which intuitively represents the average distance to the nearest neighbors of a given point.
den-SNE and densMAP not only capture additional information beyond existing visualization but also biological insights others miss; specialization of monocytes and dendritic cells; and temporally modulated transcriptomic variability.

□ scSDAEs: Sparsity-Penalized Stacked Denoising Autoencoders for Imputing Single-Cell RNA-Seq Data
>> https://www.mdpi.com/2073-4425/11/5/532
scSDAEs on recovering the true values of gene expression and helping downstream analysis, and can recover the true values and the sample–sample correlations of bulk sequencing data with simulated noise.
scSDAEs adopt stacked denoising autoencoders with a sparsity penalty, as well as a layer-wise pretraining procedure to improve model fitting. scSDAEs can capture nonlinear relationships among the data and incorporate information about the observed zeros.
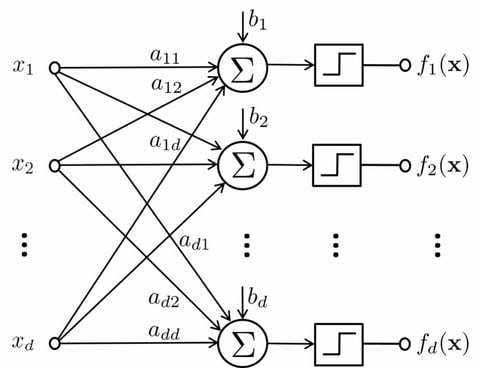
□ PALLAS: Penalized mAximum LikeLihood and pArticle Swarms for inference of gene regulatory networks from time series data
>> https://www.biorxiv.org/content/10.1101/2020.05.13.093674v1.full.pdf
PALLAS is based on the Partially-Observable Boolean Dynamical System (POBDS) model and thus does not require ad-hoc binarization of the data. The penalty in the likelihood is a LASSO regularization term, which encourages the resulting network to be sparse.
PALLAS is able to scale to large networks under no prior knowledge, by virtue of a novel continuous-discrete particle swarm algorithm for efficient simultaneous maximization of the penalized likelihood over the discrete space and the continuous space of observational parameters.

□ APEC: an accesson-based method for single-cell chromatin accessibility analysis
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02034-y
APEC (an accessibility pattern-based epigenomic clustering), which classifies each cell by groups of accessible regions with synergistic signal patterns termed “accessons”.
APEC can perform fine cell type clustering on single cell chromatin accessibility data. It can also be used to evaluate gene expression from relevant accesson, search for differential motifs/genes for each cell cluster, find super enhancers, and construct pseudo-time trajectory.
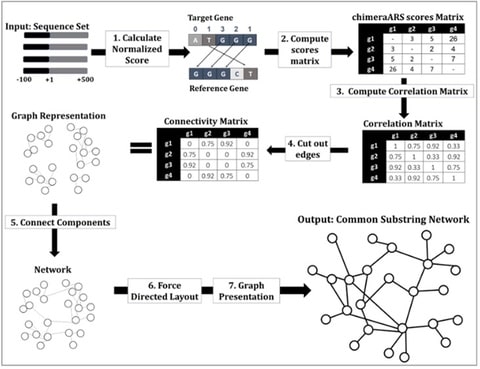
□ CSN: unsupervised approach for inferring biological networks based on the genome alone
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-3479-9
Common Substring Network (CSN) algorithm enables inferring novel regulatory relations among genes based only on the genomic sequence of a given organism and partial homolog/ortholog-based functional annotation.
CSN algorithm first calculates the common-chimeraARS scores for all pairs of genomic sequences. The pipeline can be easily improved and generalized in various dimensions and directions.

□ DeepSort: Reference-free Cell-type Annotation for Single-cell Transcriptomics using Deep Learning with a Weighted Graph Neural Network
>> https://www.biorxiv.org/content/10.1101/2020.05.13.094953v1.full.pdf
DeepSort using a modified graph neural network (GNN) model. DeepSort was constructed based on the weighted GNN framework and was then learned in two embedded high-quality scRNA-seq atlases.
DeepSort architecture consists of: the embedding layer, the weighted graph aggregator layer and the linear classifier layer. The weighted graph aggregator layer uses inductive learning to ascertain graph structure information; GraphSAGE was applied as the backbone GNN framework.
a self-loop confidence was added to the weighted graph for each cell node, and generates a linear separable feature space for cells. The final linear classifier layer classifies the final cell state representation into one of the predefined cell type categories.
□ GREMA: Modelling of emulated gene regulatory networks with confidence levels based on evolutionary intelligence to cope with the underdetermined problem
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa267/5836502
GREMA (Gene networks Reconstruction using Evolutionary Modelling Algorithm) is a program for inferring a novel type of gene regulatory network (GRN) with confidence levels for every inferred regulation, which is emulated GRN.
The higher the confidence level, the more accurate the inferred regulation. GREMA gradually determines the regulations of an eGRN with confidence levels in descending order using either an S-system or a Hill function-based ordinary differential equation model.
□ iSOM-GSN: An Integrative Approach for Transforming Multi-omic Data into Gene Similarity Networks via Self-organizing Maps
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa500/5837105
iSOM-GSN, a systematic, generalized method used to transform “multi-omic” data with higher dimensions onto a two-dimensional grid.
Based on the idea of Kohonen’s self-organizing map, iSOM-GSN generates a two-dimensional grid for each sample for a given set of genes that represent a gene similarity network.
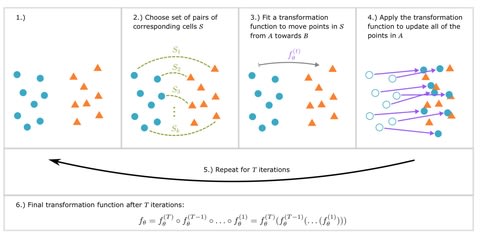
□ SCIPR: Iterative point set registration for aligning scRNA-seq data
>> https://www.biorxiv.org/content/10.1101/2020.05.13.093948v1.full.pdf
SCIPR combines many of the desirable features of previous methods including the fact that its unsupervised, generalizable, and keeps the original (gene space) representation.
When evaluating SCIPR, the local inverse Simpson’s Index (LISI) to quantify both cell type mixing and batch mixing. This leads to two values for each alignment task which can be combined for ranking the different methods by computing the difference of the medians iLISI − cLISI.
□ COTAN: Co-expression Table Analysis for scRNA-seq data
>> https://www.biorxiv.org/content/10.1101/2020.05.11.088062v1.full.pdf
COTAN provides an approximate p-value for the GPA test, and a signed co-expression index (COEX), which measures the direction and significance of the deviation from the independence hypothesis.
COTAN uses raw UMI counts, but then, for computing co-expressions these are coded as zero/non-zero.

□ CasSQ: Automated inference of Boolean models from molecular interaction maps
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa484/5836892
CaSQ by defining conversion rules and logical formulas for inferred Boolean models according to the topology and the annotations of the starting molecular interaction maps.
CaSQ is able to process large and complex maps built with CellDesigner (either following SBGN standards or not) and produce Boolean models in a standard output format, SBML-qual, that can be further analyzed.
□ GEM: Scalable and flexible gene-environment interaction analysis in millions of samples
>> https://www.biorxiv.org/content/10.1101/2020.05.13.090803v1.full.pdf
GEM (Gene-Environment interaction analysis in Millions of samples), which supports the inclusion of multiple GEI terms and adjustment for GEI covariates, conducts both model-based and robust inference procedures, and enables multi-threading to reduce computational time.

□ DCI: Learning Causal Differences between Gene Regulatory Networks
>> https://www.biorxiv.org/content/10.1101/2020.05.13.093765v1.full.pdf
The difference causal inference (DCI) algorithm infers changes (i.e., edges that appeared, disappeared or changed weight) between two causal graphs given gene expression data from the two conditions.
DCI algorithm is efficient in its use of samples and computation since it infers the differences between causal graphs directly without estimating each possibly large causal graph separately.
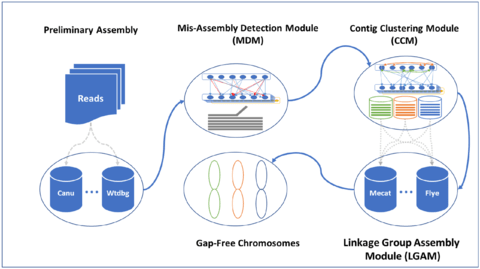
□ GALA: gap-free chromosome-scale assembly with long reads
>> https://www.biorxiv.org/content/10.1101/2020.05.15.097428v1.full.pdf
GALA by gap-free and chromosome-scale assemblies of Pacbio or Nanopore sequencing data from two publicly available datasets where the original assembly contains large gaps and a number of unanchored scaffolds.
GALA identifies multiple linkage groups, each representing a single chromosome, and describing chromosome structure with raw reads and assembled contigs from multiple de novo assembly, assembly of each linkage group by integrating results and inferring from the raw reads.
a mis-assembly detection module is achieved through cutting out the contradictory cross-layer edges. the contig-clustering module pools the linked nodes within different layers and those inside the same layer into different linkage groups, usually each representing a chromosome.

□ scVAE: Variational auto-encoders for single-cell gene expression data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa293/5838187
scVAE has support for several count likelihood functions and a variant of the variational auto-encoder has a priori clustering in the latent space.
scVAE framework for directly modelling raw counts from RNA-seq data; Gaussian-mixture VAE (GMVAE) learns biologically plausible groupings of higher adjusted Rand index.
pθ (x|z) for VAE uses non-linear transformations, posterior probability distribution pθ(z|x)=pθ(x|z)pθ(z)/pθ(x) becomes intractable.
GM-VAE has added complexity:
L(θ, φ; x) = [Eqφ(y|x)Eqφ(z|x,y) [log pθ(x|z)]
−KL(qφ(z|x)||pθ(z|y)]
−KL(qφ(y|x)||pθ(y))
□ DeepTE: a computational method for de novo classification of transposons with convolutional neural network
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa519/5838183
DeepTE utilized co-occurrence of k-mers towards TE sequences as input vector, and seven k-mer size was testified to be suitable for the classification. Eight models have been trained for different TE classification. DeepTE applied domains from TEs to correct false classification.

□ Benchmarking atlas-level data integration in single-cell genomics
>> https://www.biorxiv.org/content/10.1101/2020.05.22.111161v1.full.pdf
These real data represent complex, nested batch-effect scenarios; therefore, careful assessment of the “ground truth” is required. This simulation tasks allowed us to assess the integration methods in a setting where the nature of the batch effect could be determined and the ground truth is known.
The deep learning (DL) methods, scVI and trVAE, performed better with increasing cell numbers and batch complexity. scVI performed particularly well when the task contained complex batch effects (e.g., microwell-seq, single-cell and single-nuclei, or scATAC-seq data) and sufficient numbers of cells were present to fit these effects.
benchmarking atlas-level data integration tools: MNN , Seurat v3 , scVI , Scanorama , batch-balanced k-nearest neighbors (BBKNN) , LIGER , Conos, Harmony, a bulk data integration tool (ComBat), and a perturbation modeling tool [transformer variational autoencoder (trVAE)].

□ INSCT: Integrating millions of single cells using batch-aware triplet neural networks
>> https://www.biorxiv.org/content/10.1101/2020.05.16.100024v1.full.pdf
INSCT (“Insight”), a novel deep learning algorithm to overcome batch effects using batch-aware triplet neural networks, generates an embedding space which accurately integrates cells across experiments, platforms and species.

□ Exact-RFS-2: Advancing Divide-and-Conquer Phylogeny Estimation
>> https://www.biorxiv.org/content/10.1101/2020.05.16.099895v1.full.pdf
Exact-RFS-2, the polynomial-time algorithm to find an optimal supertree of two trees, using the Robinson-Foulds Supertree. GreedyRFS is a greedy heuristic that operates by repeatedly using Exact-RFS-2 on pairs of trees, until all the trees are merged into a single supertree.

□ Kmer2SNP: reference-free SNP calling from raw reads based on matching
>> https://www.biorxiv.org/content/10.1101/2020.05.17.100305v1.full.pdf
Kmer2SNP computes the maximum weight matching in the above heterozygous k-mer graph, where the maximum weight matching is a set of pairwise non-adjacent edges in which the sum of weights is maximized.
□ ASHURE: A workflow for accurate metabarcoding using nanopore MinION sequencing
>> https://www.biorxiv.org/content/10.1101/2020.05.21.108852v1.full.pdf
ASHURE is not limited to RCA data, as it performs a search for primers in the sequence data, splits the reads at primer binding sites, and stores the information on start and stop location of the fragment as well as its orientation.
ASHURE mitigates the high error rates associated with nanopore-based long-read single-molecule sequencing by using rolling circle amplification with a subsequent assembly of consensus sequences leading to a median accuracy of up to 99.3% for long RCA fragments.

□ Perler: Model-based prediction of spatial gene expression via generative linear mapping
>> https://www.biorxiv.org/content/10.1101/2020.05.21.107847v1.full.pdf
Perler estimates a generative linear model-based mapping function that transforms ISH data into the scRNA-seq space, thereby enabling calculation of pairwise distances between ISH data and scRNA-seq data by EM algorithm.
Perler reconstructs spatial gene- expression profiles according to the weighted mean of scRNA-seq data, which is optimized by the mapping function. a gene-expression vector for each cell in a given tissue sample measured by ISH can be mapped to the scRNA-seq space.


□ BatchBench: Flexible comparison of batch correction methods for single-cell RNA-seq
>> https://www.biorxiv.org/content/10.1101/2020.05.22.111211v1.full.pdf
BatchBench, a modular and flexible pipeline for comparing batch correction methods for single-cell RNA-seq data.
BatchBench evaluates batch correction methods based on two different entropy metrics. the entropies are not suitable for evaluating its performance as the method operates by identifying nearest neighbours in each of the provided batches and adjusting neighbors to maximize the batch entropy.

□ scDBM: Generating Synthetic Single-Cell RNA-Sequencing Data from Small Pilot Studies using Deep Learning
>> https://www.biorxiv.org/content/10.1101/2020.05.27.119594v1.full.pdf
Deep generative models are promising for sample size determination as they learn important parts of the correlation structure from a subsequently generate synthetic data from varying numbers of cells for evaluation of cluster stability in the envisioned data analysis workflow.
A single-cell deep Boltzmann machines (scDBM) outperform scVI. scDBM employs the exponential family harmonium framework that allows restricted Boltzmann machines (RBMs), the single-hidden layer version of DBMs, to deal with any distribution.

□ A Bayesian data fusion based approach for learning genome-wide transcriptional regulatory networks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-3510-1
a data fusion approach for learning transcriptional Bayesian Networks in a high-dimensional space, exploiting heterogeneous omics-data integration, to determine the transcriptional architecture.
This multi-layered -omics data integration can reveal topological hierarchies as a reflection of the transcriptional impact on gene regulation, which, to our knowledge, have not been investigated with a Bayesian learning strategy on a genomic scale.
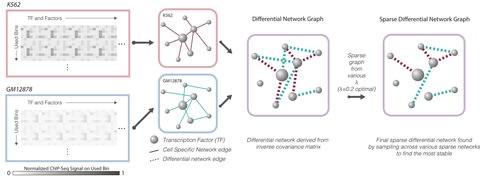
□ DiNeR: a Differential Graphical Model for analysis of co-regulation Network Rewiring
>> https://www.biorxiv.org/content/10.1101/2020.05.29.124164v1.full.pdf
DiNeR, a TF-TF network rewiring and regulator prioritization method by applying non- parametric graphical models on large-scale functional genomics data.
DiNeR uses the Gaussian graphical model (GGM) to capture the gained and lost edges in the co-regulation network. Differential network derived from inverse covariance matrix. Final sparse differential network found by sampling across various sparse networks to find the most stable.
□ MINTyper: A method for generating phylogenetic distance matrices with long read sequencing data
>> https://www.biorxiv.org/content/10.1101/2020.05.28.121251v1.full.pdf
By employing automated reference identification, KMA alignment, optional methylation masking, recombination SNP pruning and pairwise distance calculations.
MINTyper builds a complete pipeline for rapidly and accurately calculating the phylogenetic distances between a set of sequenced isolates with a presumed epidemiolocigal relation.

□ NIMBus: a Negative Binomial Regression based Integrative Method for Mutation Burden Analysis
>> https://www.biorxiv.org/content/10.1101/2020.05.29.124149v1.full.pdf
NIMBus using a Gamma-Poisson mixture model to capture the mutation-rate heterogeneity across different individuals and estimating regional background mutation rates by regressing the varying local mutation counts against genomic features extracted from ENCODE.
NIMBus automatically utilizes the genomic regions with the highest credibility for training purposes, so users do not have to be concerned about performing carefully calibrated training data selection and complex covariate matching processes.

□ RefShannon: A genome-guided transcriptome assembler using sparse flow decomposition
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0232946
RefShannon exploits the varying abundances of the different transcripts, in enabling an accurate reconstruction of the transcripts.
RefShannon using the sparse flow decomposition algorithm that was initially proposed in the Shannon assembler, which applies linear programming to efficiently decompose for the minimum number of paths at each node restricted by the node’s in-edge and out-edge weights.
□ FIRM: Fast Integration of single-cell RNA-sequencing data across Multiple platforms
>> https://www.biorxiv.org/content/10.1101/2020.06.02.129031v1.full.pdf
FIRM not only generates robust integrated datasets for downstream analysis, but is also a facile way to transfer cell type labels and annotations from one dataset to another, making it a versatile and indispensable tool for scRNA-seq analysis.
FIRM harmonizes datasets using a re-scaling procedure without modifying the underlying expression data for each cell separately, so that the relative expression patterns across cells within each dataset can be largely preserved.

□ MINTIE: identifying novel structural and splice variants in transcriptomes using RNA-seq data
>> https://www.biorxiv.org/content/10.1101/2020.06.03.131532v1.full.pdf
MINTIE combines de novo assembly of transcripts with differential expression analysis, to identify up-regulated novel variants in a case sample. MINTIE can detect any kind of anomalous sequence insertion/deletion or splicing in any gene.
MINTIE utilises de novo transcriptome assembly to reconstruct transcript sequences. It has the advantage of allowing complex variants to be detected. MINTIE can only identify structural rearrangements that affect transcribed regions.





※コメント投稿者のブログIDはブログ作成者のみに通知されます