









(Art by JT DiMartile)

□ GeneTrajectory: Gene trajectory inference for single-cell data by optimal transport metrics
>> https://www.nature.com/articles/s41587-024-02186-3
GeneTrajectory, an approach that identifies trajectories of genes rather than trajectories of cells. Specifically, optimal transport distances are calculated between gene distributions across the cell–cell graph to extract gene programs and define their gene pseudotemporal order.
Gene Trajectory provides a "movie-like" perspective to visualize how different biological processes are coordinating and governing different cell populations. Sequential trajectory identification using a diffusion-based strategy.
The initial node (terminus-1) is defined by the gene with the largest distance from the origin in the Diffusion Map embedding. GeneTrajectory then employs a random-walk procedure to select the other genes that belong to the trajectory terminated at terminus-1.
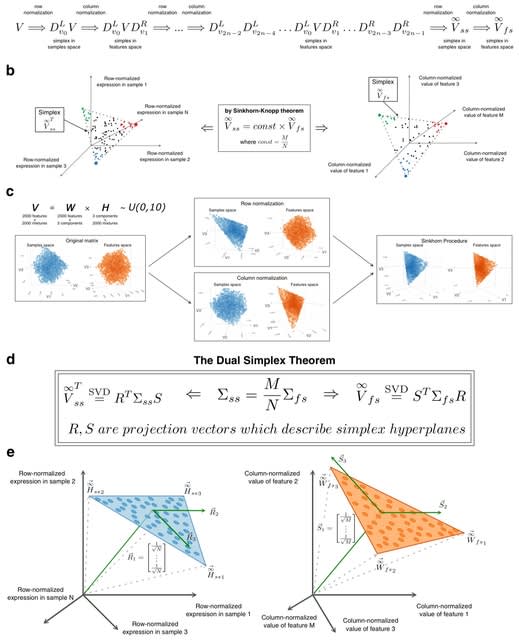
□ Non-negative matrix factorization and deconvolution as dual simplex problem
>>
https://www.biorxiv.org/content/10.1101/2024.04.09.588652v1
An analytical framework that reveals dual/complementary simplexes within the features and samples spaces. This can be achieved analytically by using projective formulation of the factorization/deconvolution problem for the Sinkhorn transformed non-negative matrix.
Sinkhorn transformation is a process of iterative multiplication by diagonal matrices, producing two converging sequences of matrices. Singular vectors of Sinkhorn-transformed matrices provide projection vectors to hyperplanes in which samples and features simplexes a located.
Dual simplex problem is equivalent to problem of finding single simplex with constraint on its inverse. The dramatic reduction in the number of optimized variables achieved by Dual Simplex approach. Gradient descent computation achieves the minimal formulation of the Dual Simplex problem.

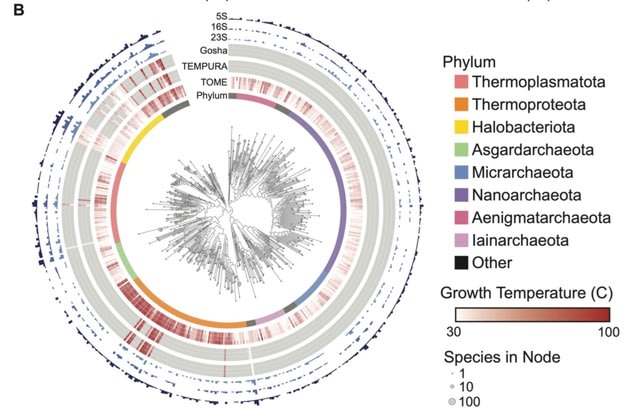
□ GARNET: RNA language models predict mutations that improve RNA function
>> https://www.biorxiv.org/content/10.1101/2024.04.05.588317v1
GARNET (Gtdb Acquired RNa with Environmental Temperatures), a new database for RNA structural analysis anchored to the GTDB. GARNET links RNA sequences derived from GTDB genomes to experimental and predicted optimal growth temperatures of GTDB reference organisms.
GARNET can define the minimal requirements for a sequence- and structure-aware RNA generative model. They also develop a GPT-like language model for RNA in which triplet tokenization provides optimal encoding.
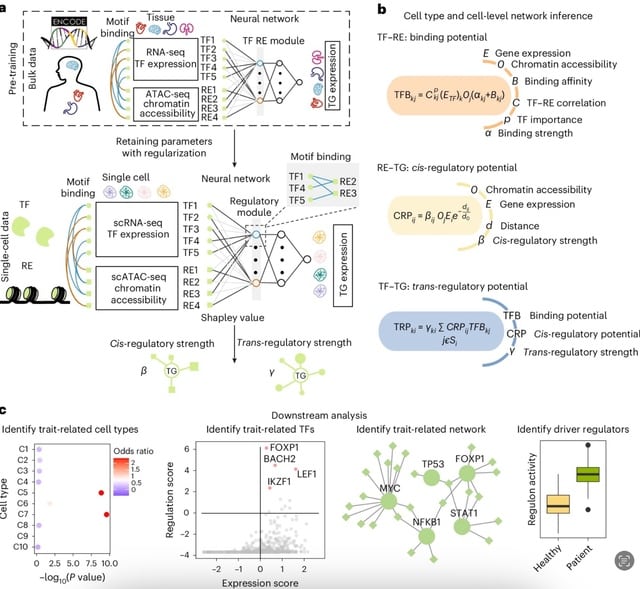
□ LINGER: Inferring gene regulatory networks from single-cell multiome data using atlas-scale external data
>> https://www.nature.com/articles/s41587-024-02182-7
LINGER leverages external data to enhance the inference from single-cell multiome data, incorporating three key steps: training on external bulk data, refining on single-cell data and extracting regulatory information using interpretable artificial intelligence techniques.
LINGER uses lifelong learning, a previously defined concept that incorporates large-scale external bulk data, mitigating the challenge of limited data but extensive parameters. LINGER integrates TF–RE motif matching knowledge through manifold regularization.
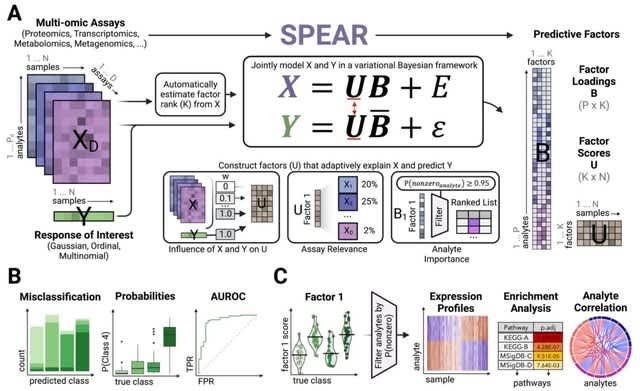
□ SPEAR: A supervised bayesian factor model for the identification of multi-omics signatures
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae202/7644285
SPEAR (Signature-based multiPle-omics intEgration via lAtent factoRs) employs a probabilistic Bayesian framework to jointly model multi-omics data with response(s) of interest, emphasizing the construction of predictive multi-omics factors.
SPEAR adaptively determines factor rank, emphasis on factor structure, data relevance and feature sparsity. SPEAR estimates analyte significance per factor, extracting the top contributing analytes as a signature.
The SPEAR model is amenable to various types of responses in both regression and classification tasks, permitting both continuous responses such as antibody titer and gene expression values, as well as categorical responses like disease subtypes.
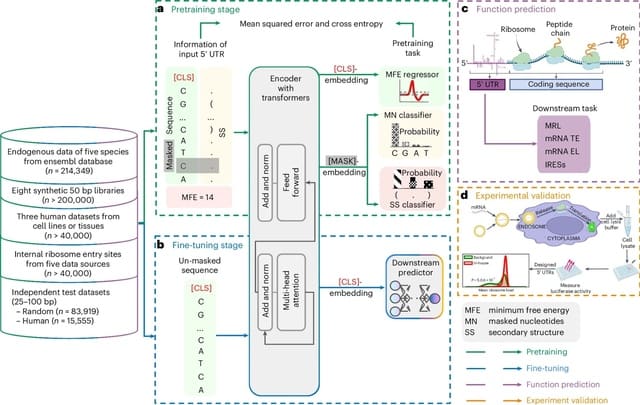
□ UTR-LM: A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions
>> https://www.nature.com/articles/s42256-024-00823-9
UTR-LM, a language model for 5′ UTR is pretrained on endogenous 5′ UTRs from multiple species and is further augmented with supervised information including secondary structure and minimum free energy.
In the UTR-LM model, the input of the pre-trained model is the 5' UTR sequence, which is fed into the transformer layer through a randomly generated 128-dimensional embedding for each nucleotide and a special [CLS] token.
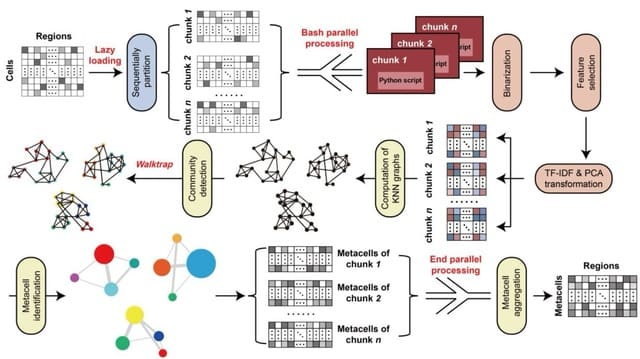
□ EpiCarousel: memory- and time-efficient identification of metacells for atlas-level single-cell chromatin accessibility data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae191/7642398
EpiCarousel is sufficient to analyze the atlas-level dataset with over 700 thousand cells and 1 million peaks using efficient RAM consumption (under 75 GB) within 2 hours, enabling users to analyze large-scale datasets on low-cost devices.
The output metacell-by-region matrix can be seamlessly integrated into the scCAS data analysis pipelines, facilitating in-depth investigation. Given a scCAS data count matrix stored in the compressed sparse row format, EpiCarousel generates a metacell-by-region/peak matrix.
EpiCarousel loads the scCAS dataset and partitions it into multiple chunks, then performs data preprocessing and identifies metacells for each chunk in parallel, and finally combines the metacells derived from each chunk to facilitate diverse downstream analyses.
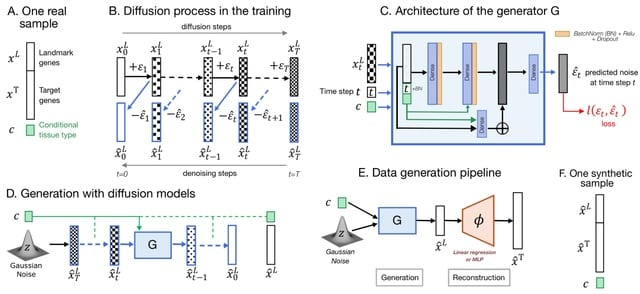
□ In Silico Generation of Gene Expression profiles using Diffusion Models
>> https://www.biorxiv.org/content/10.1101/2024.04.10.588825v1
The DDIM is trained with many epochs 15,000 due to numerous diffusion steps 1,000 and a more expressive architecture. They adapted the architecture as a residual block of the same input and output size for they could not use the typical U-NET model.
Diffusion Models also leverage the power of attention mechanisms and sophisticated class conditioning. They used Automatic Mixed Precision alongside a learning rate warmup strategy and big batch sizes to keep an efficient training time.
In addition to the residual block lavers dimensions and the learning rate, They optimized the dropout rate, the variance (Bt) scheduler (constant, linear, or quadratic), and the conditioning time steps with or without sinusoidal embedding.

□ scRCA: a Siamese network-based pipeline for the annotation of cell types using imperfect single-cell RNA-seq reference data
>> https://biorxiv.org/cgi/content/short/2024.04.08.588510v1
scRCA is the first deep-learning-based computational pipeline which is dedicated to cell type annotation using reference datasets containing noise. To improve the model's interpretability, scRCA uses an "interpreter', which defines marker genes required to classify cell types.
ScRCA employs categorical cross-entropy (CCE) as the loss function. They employed other loss functions: CCE loss, FW (forward) loss, DMI (determinant-based mutual information) loss, and generalized cross-entropy loss (GCE) loss, to implement four benchmarking methods of scRNA.

□ Annotatability: Interpreting single-cell and spatial omics data using deep networks training dynamics
>> https://www.biorxiv.org/content/10.1101/2024.04.06.588373v1
Annotatability, a framework for annotation-trainability analysis, achieved by monitoring the training dynamics of deep neural networks. Annotatability improves the single-cell genomics annotations, identifies intermediate cell states, and enables signal-aware downstream analysis.
Anotatability is equipped with a training-dynamics-based score that captures either positive or negative association of genes relative to a given biological signal, revealed by their correlation or anti-correlation with the confidence in a particular annotation.
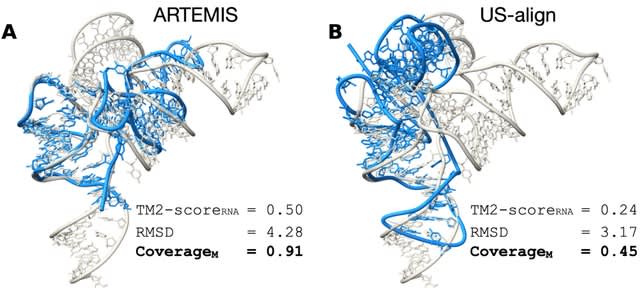
□ ARTEMIS: a method for topology-independent superposition of RNA 3D structures and structure-based sequence alignment
>> https://www.biorxiv.org/content/10.1101/2024.04.06.588371v1
ARTEMIS operates in polynomial time and ensures the optimal solution, provided it includes at least one residue-residue match with a near-zero RMSD. ARTEMIS significantly outperforms SOTA tools in both sequentially-ordered and topology-independent RNA 3D structure superposition.
Leveraging ARTEMIS, they discovered a helical packing motif to be preserved in different backbone topology contexts in diverse non-coding RNAs, including multiple ribozymes and riboswitches.
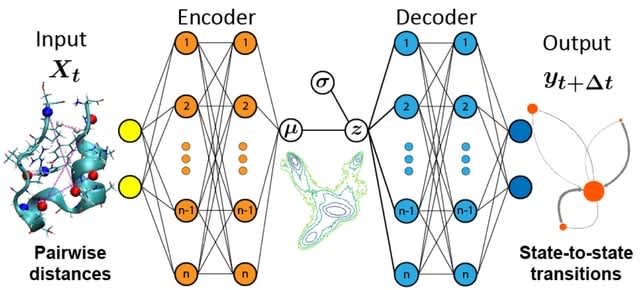
□ An Information Bottleneck Approach for Markov Model Construction
>> https://arxiv.org/abs/2404.02856
Constructing the Markovian model at a specific lag time requires state defined without significant internal energy barriers, enabling internal dynamics relaxation w/in the lag time. This process coarse grains time and space, integrating out rapid motions within metastable states.
A continuous embedding approach for molecular conformations using the state predictive information bottleneck (SPIB), which unifies dimensionality reduction and state space partitioning via a continuous, machine learned basis set.
SPIB dentifies slow dynamical processes and constructing predictive multi-resolution Markovian models. SPIB showcases unique advantages compared to competing methods. It automatically adjusts the number of metastable states based on a specified minimal time resolution.

□ COVET / ENVI: The covariance environment defines cellular niches for spatial inference
>> https://www.nature.com/articles/s41587-024-02193-4
COVET, a compact representation of a cell’s niche that assumes that interactions between the cell and its environment create biologically meaningful covariate structure in gene expression between cells of the niche.
COVET uses a corresponding distance metric that unlocks the ability to compare and analyze niches using the full toolkit of approaches currently employed for cellular phenotypes, including dimensionality reduction, spatial gradient analysis and clustering.
ENVI (environmental variational inference), a conditional variational autoencoder (CVAE) simultaneously incorporates scRNA-seq and spatial data into a single embedding.
ENVI leverages the covariate structure of COVET as a representation of cell microenvironment and achieves total integration by encoding both genome-wide expression and spatial context (the ability to reconstruct COVET matrices) into its latent embedding.

□ Pantera: Identification of transposable element families from pangenome polymorphisms
>> https://www.biorxiv.org/content/10.1101/2024.04.05.588311v1
A pangenome is a collection of genomes or haplotypes that can be aligned and stored as a variation graph in gfa format. pantera receives as input a list of gfa files of non overlapping variation graphs and produces a library of transposable elements found to be polymorphic on that pangenome.
Pantera selects from the gfa file segments that are polymorphic. To reduce the FP only segments for which there are at least two identical polymorphic sequences are selected. Then, a less stringent clustering is performed to reduce redundancy and generate the final TE library.
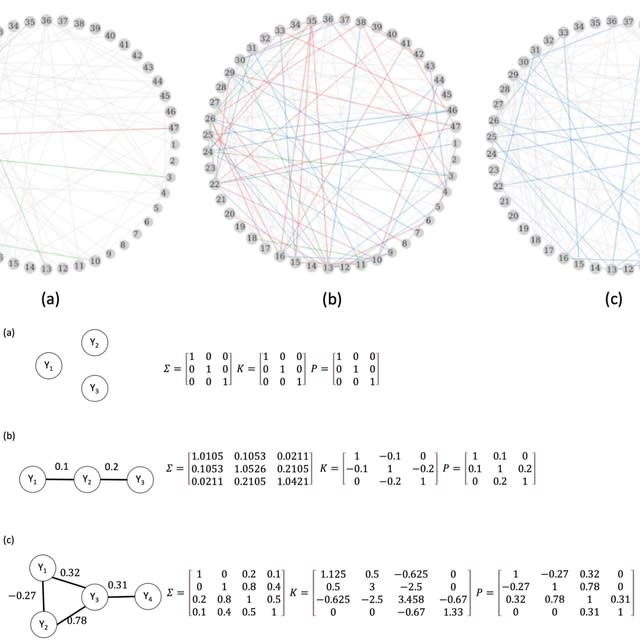
□ Learning Gaussian Graphical Models from Correlated Data
>> https://www.biorxiv.org/content/10.1101/2024.04.03.587948v1
A Bootstrap algorithm to learn a GGM from correlated data. The advantage of this method is that there is no need to estimate the correlations within the clusters, and the approach is not limited to family-based data. This algorithm controls the Type I error well.
A Gaussian Graphic Model (GGM) is a statistical model that represents properties of marginal and conditional independencies of a multivariate Gaussian distribution using an undirected Markov graph.
The key rule of an undirected Markov graph is that two variables are conditionally independent given all the other variables in the graph if they are not connected by an edge.

□ seqspec: A machine-readable specification for genomics assays
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae168/7641535
seqspec, a machine-readable specification for libraries produced by genomics assays that facilitates standardization of preprocessing and enables tracking and comparison of genomics assays.
Sequencing libraries are constructed by combining Atomic Regions to form an adapter-insert-adapter construct. The seqspec for the assay annotates the construct with Regions and meta Regions.
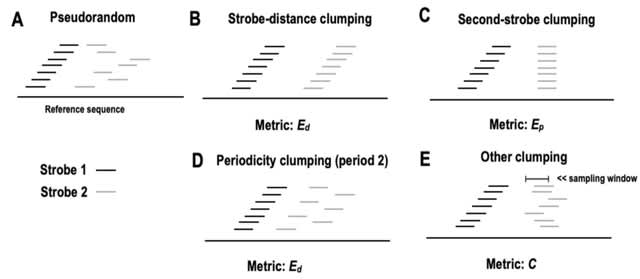
□ Designing efficient randstrobes for sequence similarity analyses
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae187/7641534
A novel construction methods, including a Binary Search Tree (BST)-based approach that improves time complexity over previous methods. They are also the first to address biases in construction and design three metrics for measuring bias.
Thier methods change the seed construction in strobealign, a short-read mapper, and find that the results change substantially. They suggest combining the two results to improve strobealign's accuracy for the shortest reads in our evaluated datasets.
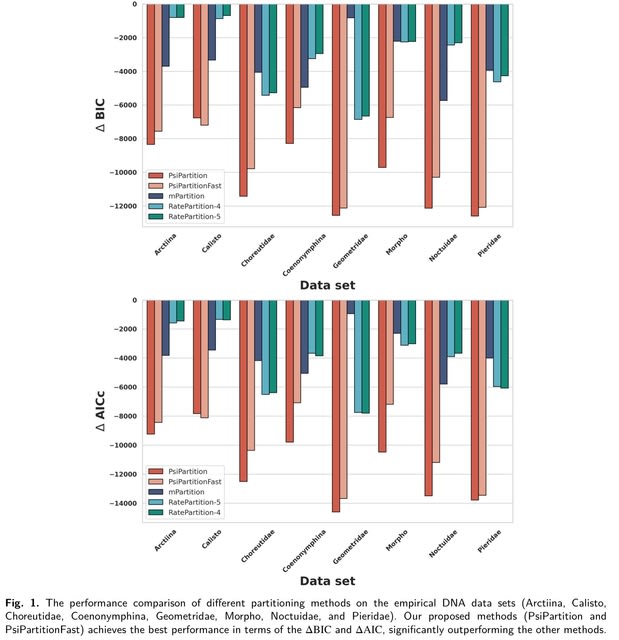
□ PsiPartition: Improved Site Partitioning for Genomic Data by Parameterized Sorting Indices and Bayesian Optimization
>> https://www.biorxiv.org/content/10.1101/2024.04.03.588030v1
PsiPartition, a novel partitioning approach based on the parameterized sorting indices of sites and Bayesian optimization.
PsiPartition evidently outperforms other methods in terms of the Robinson-Foulds (RF) distance between the true simulated trees and the reconstructed trees. It provides a new general framework to efficiently determine the optimal number of partitions.

□ VarChat: the generative AI assistant for the interpretation of human genomic variations
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae183/7641533
VarChat requires as input genomic variants coordinates according to HGVS nomenclature together with gene symbols, or to dbSNP identifier. For every queried variant, VarChat produces concise and coherent summaries through an LLM model.
VarChat enables clinicians to capture the core insights of articles associated with these variants. VarChat provides the user with the 15 most relevant references, when available. The relevance of the publication is based on a modified version of the BM25 ranking algorithm.
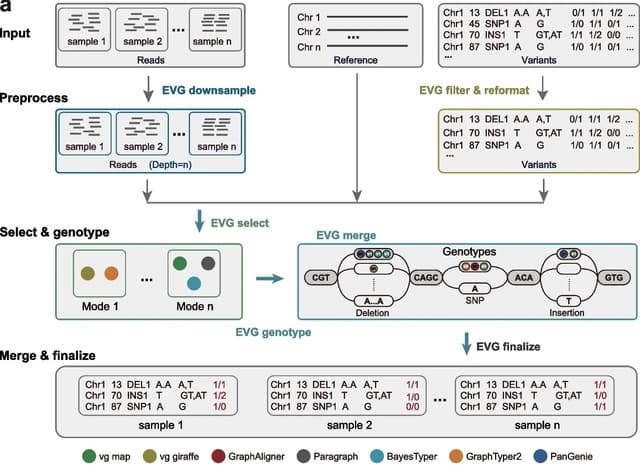
□ Ensemble Variant Genotyper: A comprehensive benchmark of graph-based genetic variant genotyping algorithms on plant genomes for creating an accurate ensemble pipeline
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03239-1
EVG (Ensemble Variant Graph-based tool) can accurately genotype SNPs, indels, and SVs using short reads. EVG achieves higher genotyping accuracy and recall with only 5× sequencing data. EVG remains robust even as the number of nodes in the pangenome graph increases.
EVG automatically selects the optimal genotyping process based on factors including the size of the reference genome, the sequencing depth of the individual genome to be genotyped, and the read length of the sequencing data.
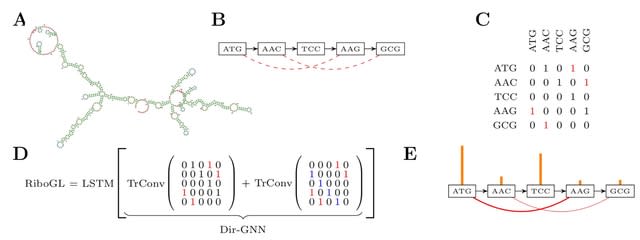
□ RiboGL: Towards improving full-length ribosome density prediction by bridging sequence and graph-based representations
>> https://www.biorxiv.org/content/10.1101/2024.04.08.588507v1
RiboGL combines graph and recurrent neural networks to account for both graph and sequence-based features. The model takes a mixed graph representing the secondary structure of the mRNA sequence as input, which incorporates both sequence and structure codon neighbors.
RiboGL uses gradient-based interpretability to understand how the codon context and the structural neighbors affect the ribosome dwell time at the A site.
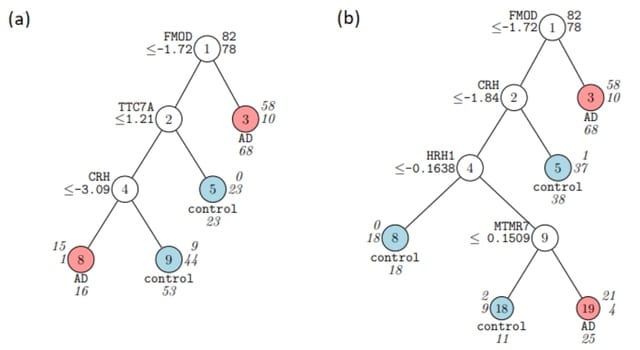
□ SIEVE: One-stop differential expression, variability, and skewness analyses using RNA-Seq data
>> https://www.biorxiv.org/content/10.1101/2024.04.09.588804v1
SIEVE adopts a compositional data analysis approach to modeling discrete RNA-Seq count data, applies Aitchison's CLR transformation to convert them into continuous form, and uses a skew-normal distribution to model them.
Subsets of the genes detected using SIEVE that are strongly predictive of the AD state were identified using the Generalized, Unbiased Interaction Detection and Estimation classification and regression tree algorithm.

□ TDEseq: Powerful and accurate detection of temporal gene expression patterns from multi-sample multi-stage single-cell transcriptomics data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03237-3
TDEseq, temporal differentially expressed genes of time-course scRNA-seq data. Specifically, TDEseq primarily builds upon a linear additive mixed model (LAMM) framework, with a random effect term to account for correlated cells within an individual.
TDEseq controls the type I error rate at the transcriptome-wide level and display powerful performance in detecting temporal expression genes under the power simulations. A linear version of TDEseq can model the small sample heterogeneity inherited in time-course scRNA-seq data.

□ slow5curl: Streamlining remote nanopore data access
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giae016/7644676
Slow5curl enables a user to extract and download a specific read or set of reads (e.g., the reads corresponding to a gene of interest) from a dataset on a remote server, avoiding the need to download the entire file.
Slow5curl uses highly parallelized data access requests to maximize speed. slow5curl can facilitate targeted reanalysis of remote nanopore cohort data, effectively removing data access as a consideration.
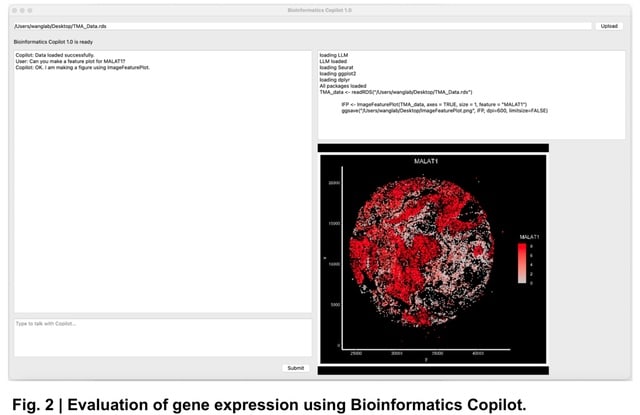
□ Bioinformatics Copilot 1.0: A Large Language Model-powered Software for the Analysis of Transcriptomic Data
>> https://www.biorxiv.org/content/10.1101/2024.04.11.588958v1
Bioinformatics Copilot 1.0, a large language model- powered software for analyzing transcriptomic data using natural language.
Bioinformatics Copilot 1.0 facilitates local data analysis, ensuring adherence to stringent data management regulations that govern the use of patient samples in medical and research institutions.
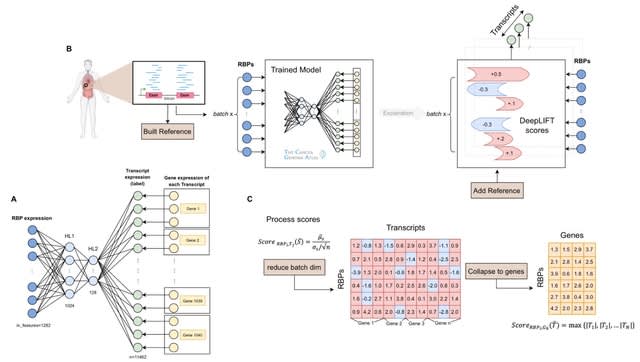
□ DeepRBP: A novel deep neural network for inferring splicing regulation
>> https://www.biorxiv.org/content/10.1101/2024.04.11.589004v1
DeepRBP, a deep learning (DL) based framework to identify potential RNA-binding proteins (RBP)-Gene regulation pairs for further in-vitro validation.
DeepRBP is composed of a DL model that predicts transcript abundance given RBP and gene expression data coupled with an explainability module that computes informative RBP- Gene scores.
□ Designing and delivering bioinformatics project-based learning in East Africa
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05680-2
EANBiT is part of the Human Heredity and Health in Africa Consortium (H3Africa) training program to develop bioinformatics and genomics expertise in Africa through postgraduate training to support the capacity building for the analysis of genomic data.

□ scPRAM accurately predicts single-cell gene expression perturbation response based on attention mechanism
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae265/7646141
sPRAM, a method for predicting Perturbation Responses in single-cell gene expression based on Attention Mechanisms. sPRAM aligns cell states before and after perturbation, followed by accurate prediction of gene expression responses to perturbations for unseen cell types.
sPRAM leverages a VAE to encode the training set into a latent space, followed by optimal transport based on Sinkhorn algorithm to pair unpaired cells. Subsequently, an attention mechanism is employed to compute perturbation vectors for test cells.

□ oHMMed: Inference of genomic landscapes using ordered Hidden Markov Models with emission densities
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05751-4
oHMMed (ordered HMM w/ emission densities) assumes continuous emissions. oHMMed provides a best-fit annotation of the observed sequence, corresponding estimates of the transition rate matrix, and estimates of the state-specific and shared parameters of the emitted distributions.
In the other, the emission density is a gamma mixture initially; however, rate parameters of poisson distributions are subsequently drawn from the individual gamma distributions, yielding an observed density of gamma-poisson mixtures where the data points are discrete counts.

□ Combining LIANA and Tensor-cell2cell to decipher cell-cell communication across multiple samples
>> https://www.cell.com/cell-reports-methods/fulltext/S2667-2375(24)00089-4
LIANA is a computational framework that implements multiple available ligand-receptor resourcesand methods to analyze CCC. Tensor-cell2cell is a dimensionality reduction approach devised to uncover context-driven CCC programs across multiple samples simultaneously. Specifically, Tensor-cell2cell uses CCC scores inferred by any method and arranges the data into a four-dimensional (4D) tensor.
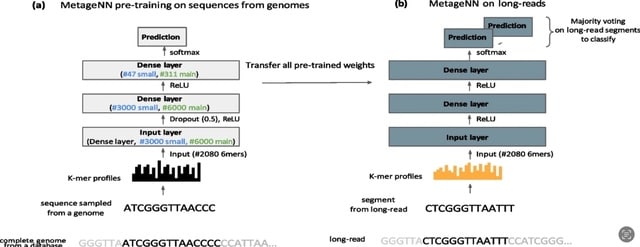
□ MetageNN: a memory-efficient neural network taxonomic classifier robust to sequencing errors and missing genomes
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05760-3
MetageNN overcomes the limitation of not having long-read sequencing-based training data for all organisms by making predictions based on k-mer profiles of sequences collected from a large genome database.
MetageNN uses short k-mer-profiles that are known to be less affected by sequencing errors to reduce the “distribution shift” between genome sequences and noisy long reads. MetageNN outperforms MetaMaps and Kraken2 in detecting potentially novel lineages.
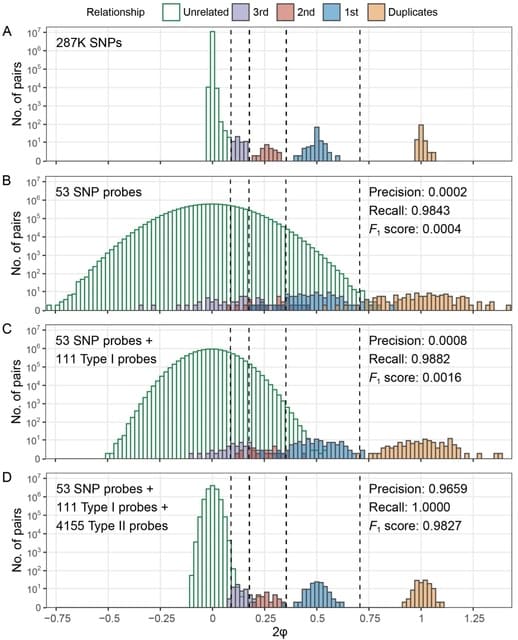
□ MethylGenotyper: Accurate estimation of SNP genotypes and genetic relatedness from DNA methylation data
>> https://www.biorxiv.org/content/10.1101/2024.04.15.589670v1
MethylGenotyper to perform genotype calling based on DNAm data for SNP probes, Type I probes, and Type II probes. For each type of probes, MethylGenotyper first converts the methylation intensity signals to the Ratio of Alternative allele Intensity (RAI).
MethylGenotyper models RAI for each type of probes with a mixture of three beta distributions and one uniform distribution, and employs an expectation-maximization (EM) algorithm to obtain the maximum likelihood estimates (MLE) of model parameters and genotype probabilities.
□ AutoGDC: A Python Package for DNA Methylation and Transcription Meta-Analyses
>> https://www.biorxiv.org/content/10.1101/2024.04.14.589445v1
AutoGDC provides the access to the Genomic Data Commons data repository, which contains more than 230,000 open-access data files and more than 350,000 controlled-access data files. The autogdc infrastructure focuses upon transcription and DNA methylation profiling data.










※コメント投稿者のブログIDはブログ作成者のみに通知されます