









(Art by JT DiMartile)
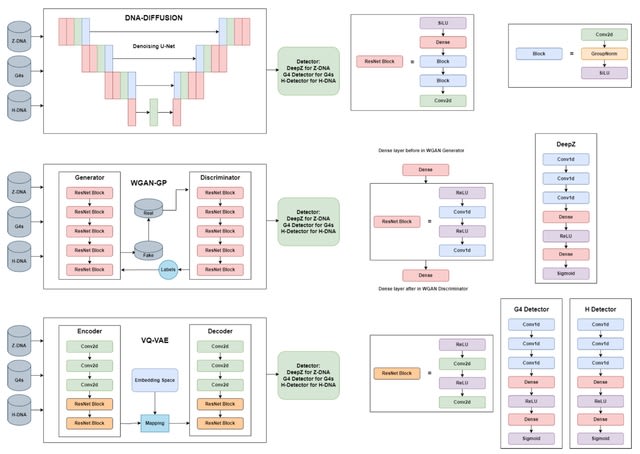
□ DNA-Diffusion: Generative Models for Prediction of Non-B DNA Structures
>> https://www.biorxiv.org/content/10.1101/2024.03.23.586408v1
VQ-VAE (Quantised-Variational AutoEncoder) learns a discrete latent variable by the encoder, since discrete representations may be a more natural fit for sequence data. Vector quantisation (VQ) is a method to map N-dimensional vectors into a finite set of latent vectors.
VQ-VAE architecture consists of encoder and decoder models. Both encoder and decoder consist of 2 strided convolutional layers with stride 2 and window size 4 x 4, followed by two residual 3 × 3 blocks (implemented as ReLU, 3x3 conv, ReLU, 1x1 conv), all having 256 hidden units.
The decoder similarly has two residual 3 x 3 blocks, followed by two transposed convolutions with the stride 2 and the window size 4 × 4. Transformation into discrete space is performed by VectorQuantizerEMA with embedding.
DNA-Diffusion generates a class of functional genomic elements - non-B DNA structures. For Z-DNA the difference is small, and both WGAN and VQ-VAE show good results. This could be due to the captured pattern of Z-DNA-prone repeats detected by Z-DNABERT attention scores.
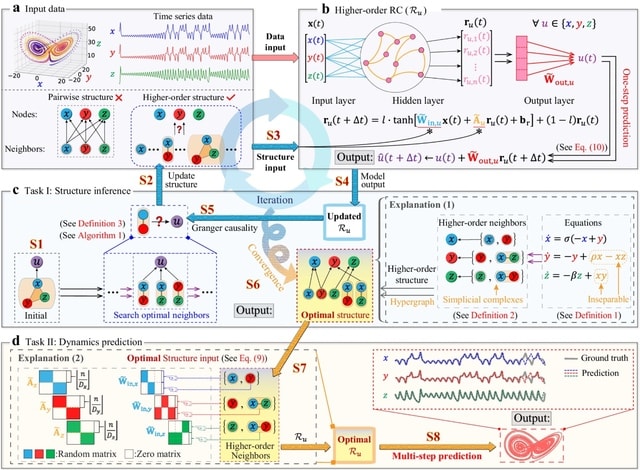
□ HoGRC: Higher-order Granger reservoir computing: simultaneously achieving scalable complex structures inference and accurate dynamics prediction
>> https://www.nature.com/articles/s41467-024-46852-1
Higher-Order Granger RC (HoGRC) first infers the higher-order structures by incorporating the idea of Granger causality into the RC. Simultaneously, HoGRC enables multi-step prediction by processing the time series along with the inferred higher-order information.
HoGRC iteratively refines the initial / coarse-grained candidate neighbors into the optimal higher-order neighbors, until an optimal structure is obtained, tending to align w/ the true higher-order structure. The GC inference and the dynamics prediction are mutually reinforcing.

□ GPTCelltype: Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis
>> https://www.nature.com/articles/s41592-024-02235-4
Assessing the performance of GPT-4, a highly potent large language model, for cell type annotation, and demonstrated that it can automatically and accurately annotate cell types by utilizing marker gene information generated from standard single-cell RNA-seq analysis pipelines.
GPTCelltype, an interface for GPT models. GPTCelltype takes marker genes or top differential genes as input, and automatically generates prompt message using the following template with the basic prompt strategy.
Using GPTCelltype as the interface, GPT-4 is also notably faster, partly due to its utilization of differential genes from the standard single-cell analysis pipelines such as Seurat.
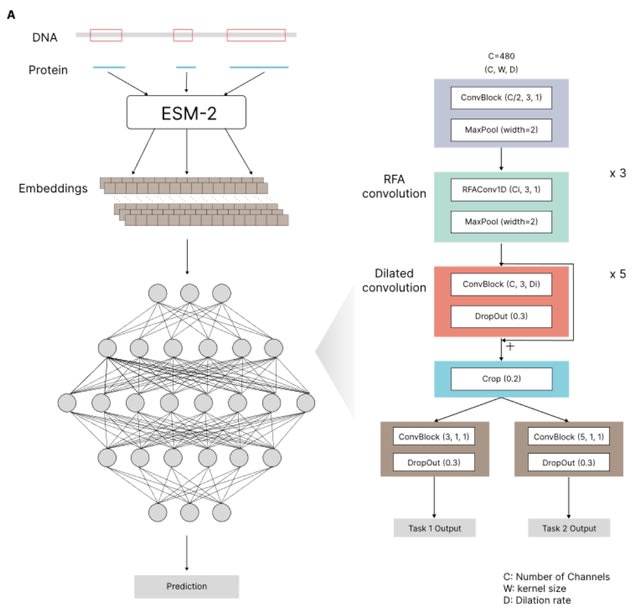
□ TraitProtNet: Deciphering the Genome for Trait Prediction with Interpretable Deep Learning
>> https://www.biorxiv.org/content/10.1101/2024.03.28.587180v1
TraitProtNet incorporates the Receptive-Field Attention (RFA) convolutional block, enabling the framework to focus on significant features through spatial attention mechanisms integrated with deep learning for advanced one-dimensional sequence data processing.
The TraitProtNet architecture comprises: four convolutional blocks with pooling to distill sequence lengths, five dilated convolutional blocks for capturing long-distance interactions between proteins, and a cropping layer that precedes two task-specific outputs.
TraitProtNet is adept at processing sequences where each element contains 480 features, utilizing max pooling to refine sequence lengths. Dynamic attention weights are generated via average pooling and depth-wise convolution, emphasizing the most informative sequence segments.
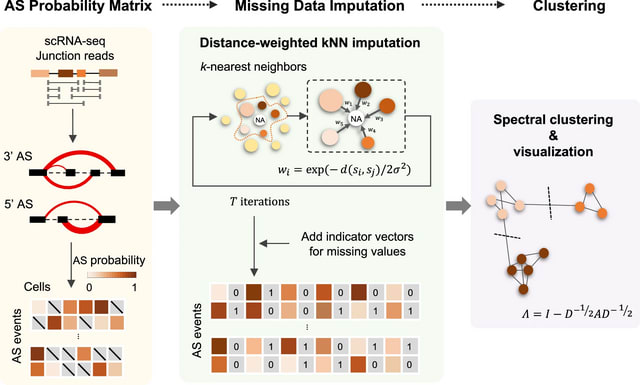
□ SCASL: Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance
>> https://www.nature.com/articles/s41467-024-46480-9
SCASL (single-cell clustering based on alternative splicing landscapes) employs a strategy similar to that of LeafCutter and FRASER to identify AS events from the junction reads in single-cell SMART-seq data. SCASL can recover both known and novel AS events.
SCASL generates classifications of cell subpopulations. SCASL introduces a strategy of iterative weighted KNN for imputation of these missing values. SCASL recovered a series of transitional stages during developments of the hepatocyte and cholangiocyte lineage lineages.
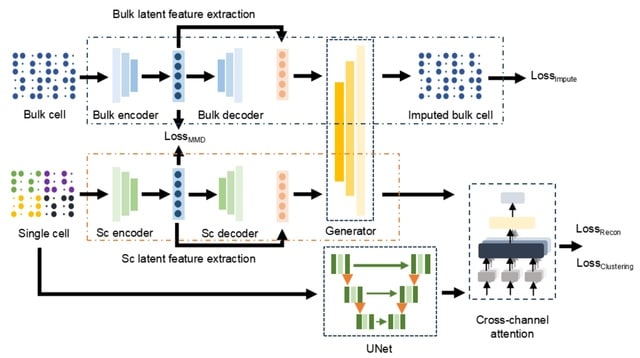
□ scDTL: single-cell RNA-seq imputation based on deep transfer learning using bulk cell information
>> https://www.biorxiv.org/content/10.1101/2024.03.20.585898v1
scDTL, a deep transfer learning based framework that addresses single-cell RNA-seq imputation problem by considering large-scale bulk RNA-seq information synchronously. scDTL firstly trains an imputation model for bulk RNA-seq data using a denoising autoencoder.
scDTL mainly consists of 1. build a imputation model via supervised learning using large-scale bulk RNA-seq data, 2. propose a framework leveraging well-trained bulk imputation model and a 1D U-net module for imputing the dropouts of a given single-cell RNA-seq expression matrix.
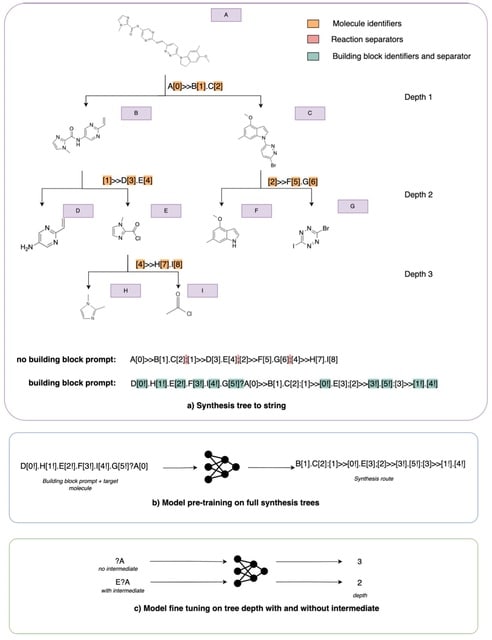
□ Leap: molecular synthesisability scoring with intermediates
>> https://arxiv.org/abs/2403.13005
Leap can adapt to available intermediates to better estimate the practical synthetic complexity of a target molecule. To create target molecule-intermediate pairs, they randomly sample a maximum of three intermediate molecules for each route and recompute the depth accordingly.
Leap computes routes using AiZynthFinder. At a synthesis tree level, Leap effectively results in the removal of any nodes beyond the intermediate molecule. This reduces the depth of the tree when the intermediate is found along the longest branch of the tree.
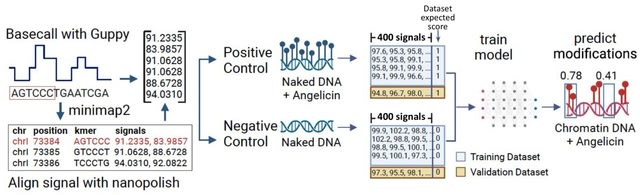
□ Probing chromatin accessibility with small molecule DNA intercalation and nanopore sequencing
>> https://www.biorxiv.org/content/10.1101/2024.03.20.585815v1
Add-seq (adduct sequencing), a method to probe chromatin accessibility by treating chromatin with the small molecule angelicin, which preferentially intercalates into DNA not bound to core nucleosomes.
Nanopore sequencing of the angelicin-modified DNA is possible and allows analysis of long single molecules w/ distinct chromatin structure. The angelicin modification can be detected from the Nanopore current signal data using a neural network model trained on chromatin-free DNA.
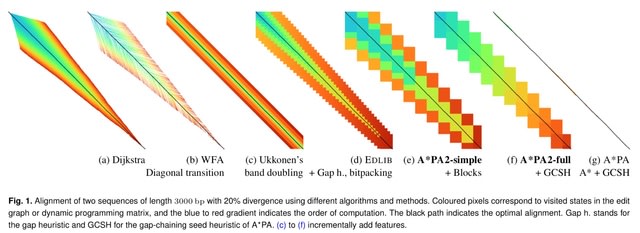
□ A*PA2: up to 20 times faster exact global alignment
>> https://www.biorxiv.org/content/10.1101/2024.03.24.586481v1
A*PA2 (Astar Pairwise Aligner 2), an exact global pairwise aligner with respect to edit distance. The goal of A*PA2 is to unify the near-linear runtime of A*PA on similar sequences with the efficiency of dynamic programming (DP) based methods.
A*PA2 uses Ukkonen's band doubling in combination with Myers' bitpacking. A*PA2 extends this with SIMD (single instruction, multiple data), and uses large block sizes inspired by BLOCK ALIGNER.
A*PA2 avoids recomputation of states where possible as suggested before by Fickett. A*PA2 introduces a new optimistic technique for traceback based on diagonal transition, and applies the heuristics developed in A*PA and improves them using pre-pruning.

□ BetaAlign: a deep learning approach for multiple sequence alignment
>> https://www.biorxiv.org/content/10.1101/2024.03.24.586462v1
BetaAlign, the first deep learning aligner, which substantially deviates from conventional algorithms of alignment computation. BetaAlign now calculates multiple alternative alignments and return the alignment that maximizes the certainty.
BetaAlign draws on natural language processing (NLP) techniques and trains transformers to map a set of unaligned biological sequences to an MSA. BetaAlign randomizes the order in which the input unaligned sequences are concatenated.

□ EXPORT: Biologically Interpretable VAE with Supervision for Transcriptomics Data Under Ordinal Perturbations
>> https://www.biorxiv.org/content/10.1101/2024.03.28.587231v1
EXPORT (EXPlainable VAE for ORdinally perturbed Transcriptomics data) an interpretable VAE model with a biological pathway informed architecture, to analyze ordinally perturbed transcriptomics data.
Specifically, the low-dimensional latent representations in EXPORT are ordinally-guided by training an auxiliary deep ordinal regressor network and explicitly modeling the ordinality in the training loss function with an additional ordinal-based cumulative link loss term.

□ MIKE: an ultrafast, assembly- and alignment-free approach for phylogenetic tree construction
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae154/7636962
MIKE (MinHash-based k-mer algorithm) is designed for the swift calculation of the Jaccard coefficient directly from raw sequencing reads and enables the construction of phylogenetic trees based on the resultant Jaccard coefficient and Mash evolutionary distances.
MIKE constructs phylogenetic trees using the computed distance matrix through the BIONJ or NJ approach. MIKE bypasses genome assembly and alignment requirements and exhibits exceptional data processing capabilities, efficiently handling large datasets in a short timeframe.
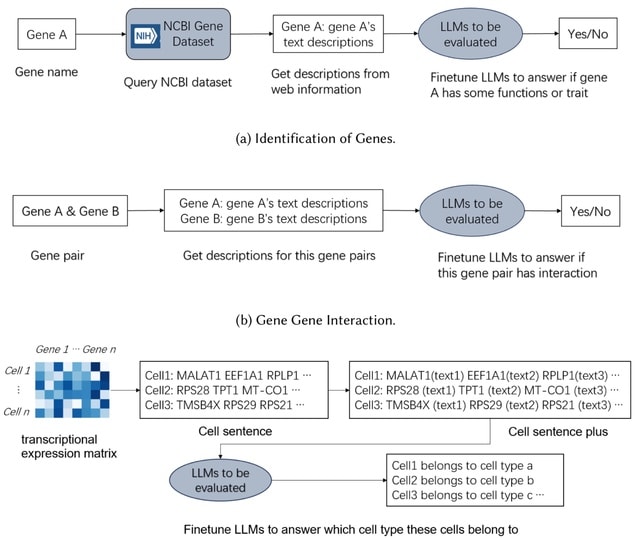
□ How do Large Language Models understand Genes and Cells
>> https://www.biorxiv.org/content/10.1101/2024.03.23.586383v1
Both GenePT and CellSentence independently employ a method where the names of the top 100 highly expressed genes are concatenated to form a textual representation of a cell, referred to as a "cell sentence".
However, they argue that such representations lack the textual structure characteristic of natural language. They have appended a succinct functional description to each gene name, a method they have dubbed "cell sentence plus".
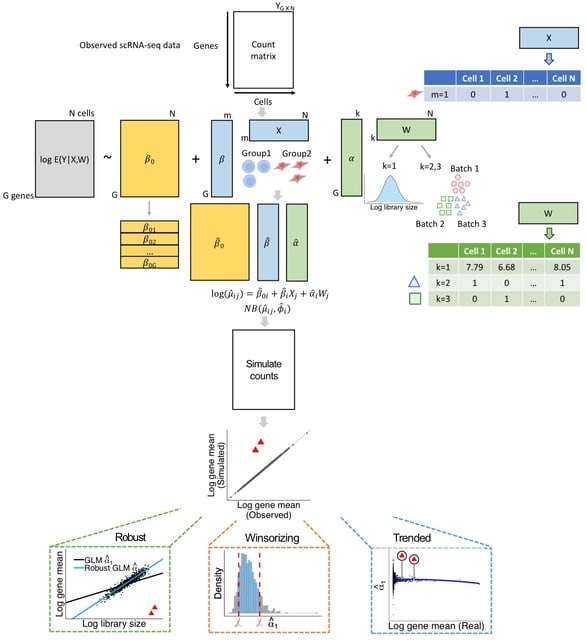
□ GLMsim: a GLM-based single cell RNA-seq simulator incorporating batch and biological effects
>> https://www.biorxiv.org/content/10.1101/2024.03.20.586030v1
GLMsim (Generalized Linear Model based simulator) fits each gene's counts into a negative binomial generalized linear model, estimating mean GE as a function of the estimated library size, biology and batch parameters and then samples counts from negative binomial distributions.
GLMsim starts from an observed scRNA-seq count matrix that includes the cell type and batch information. GLMsim captures the main characteristics of the data by fitting a generalized linear model, returning estimated parameter values for each gene.
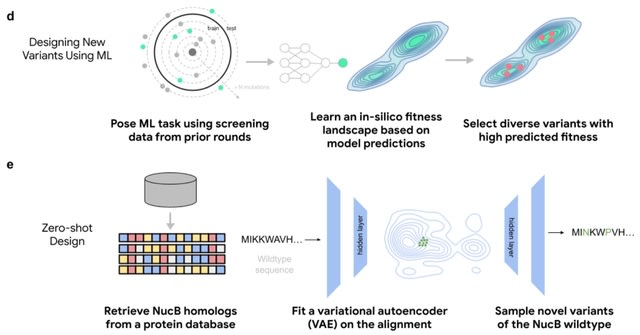
□ Engineering of highly active and diverse nuclease enzymes by combining machine learning and ultra-high-throughput screening
>> https://www.biorxiv.org/content/10.1101/2024.03.21.585615v1
ML-guided directed evolution (MLDE) can design diverse, high-activity proteins better than DE when both employ the same ultra-high-throughput microfluidics platform in a multi-round protein engineering campaign.
Engineering NucB, a biofilm-degrading endonuclease with applications in chronic wound care and anti-biofouling. NucB can degrade the DNA in the extracellular matrix required for the formation of biofilm.
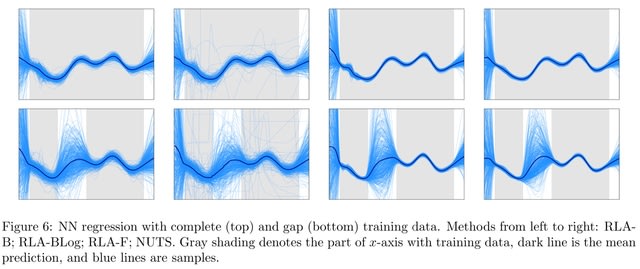
□ Riemannian Laplace Approximation with the Fisher Metric
>> https://arxiv.org/abs/2311.02766
Transforming samples from a Gaussian distribution using numerical integrators to follow geodesic paths induced by a chosen geometry, which can be carried out in parallel.
Riemann Manifold MCMC is computationally attractive due to the exponential map for transforming the samples. However, the approximation is not asymptotically exact. Fisher Information Matrix gives an approximation that is exact for targets that are diffeomorphisms of a Gaussian.
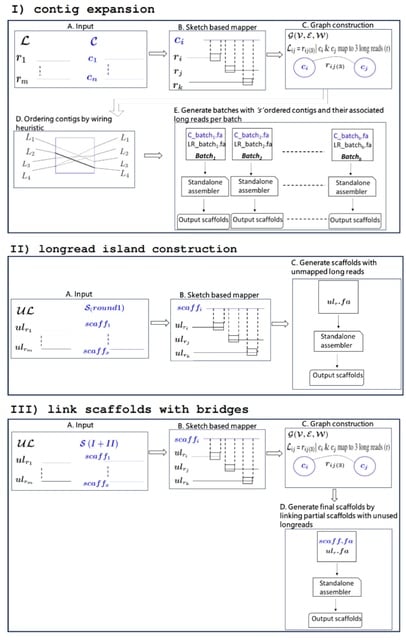
□ Maptcha: An efficient parallel workflow for hybrid genome scaffolding
>> https://www.biorxiv.org/content/10.1101/2024.03.25.586701v1
Maptcha, a new parallel workflow for hybrid genome scaffolding that would allow combining pre-constructed partial assemblies with newly sequenced long reads toward an improved assembly.
Maptcha is aimed at generating long genome scaffolds of a target genome, from two sets of input sequences---an already constructed partial assembly of contigs, and a set of newly sequenced long reads.
Maptcha internally uses an alignment-free mapping step to build a (contig,contig) graph using long reads as linking information. Subsequently, this graph is used to generate scaffolds.
□ OmicsFootPrint: a framework to integrate and interpret multi-omics data using circular images and deep neural networks
>> https://www.biorxiv.org/content/10.1101/2024.03.21.586001v1
OmicsFootPrint, a novel framework for transforming multi-omics data into two-dimensional circular images for each sample, enabling intuitive representation and analysis. OmicsFootPrint incorporates the SHapley Additive exPlanations (SHAP) algorithm for model interpretation.
The OmicsFootPrint framework can utilize various deep-learning models as its backend. A transformed circular image where data points along the circumference represent singular omics features is used as the input into the OmicsFootPrint framework for subsequent classification.
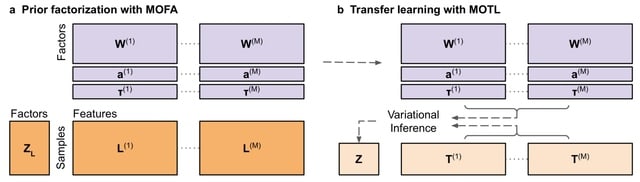
□ MOTL: enhancing multi-omics matrix factorization with transfer learning
>> https://www.biorxiv.org/content/10.1101/2024.03.22.586210v1
Joint matrix factorization disentangles underlying mixtures of biological signals, and facilitating efficient sample clustering. However, when a multi-omics dataset is generated from only a limited number of samples, the effectiveness of matrix factorization is reduced.
MOTL (Multi-Omics Transfer Learning), a novel Bayesian transfer learning algorithm for multi-omics matrix factorization. MOTL factorizes the target dataset by incorporating latent factor values already inferred from the factorization of a learning dataset.

□ GeneSqueeze: A Novel Lossless, Reference-Free Compression Algorithm for FASTQ/A Files
>> https://www.biorxiv.org/content/10.1101/2024.03.21.586111v1
GeneSqueeze, a novel reference-free compressor which uses read-reordering-based compression methodology. Notably, the reordering mechanism within GeneSqueeze is temporally confined to specific algorithmic blocks, facilitating targeted operations on the reordered sequences.
GeneSqueeze presents a dynamic protocol for maximally compressing diverse FASTQ/A files containing any IUPAC nucleotides while maintaining complete data integrity of all components.
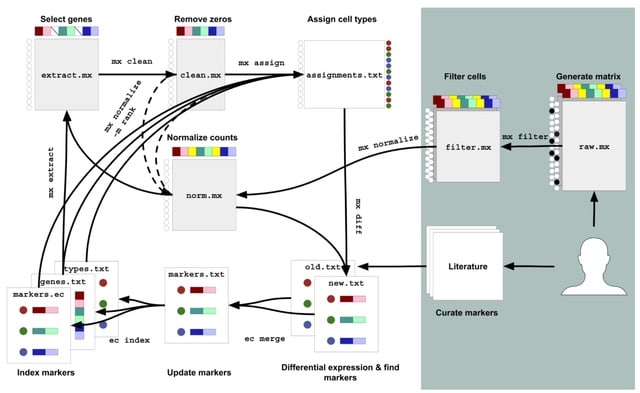
□ Algorithms for a Commons Cell Atlas
>> https://www.biorxiv.org/content/10.1101/2024.03.23.586413v1
Mx assign takes in a single-cell matrix and a marker gene file and performs cell-type assignment using a modified Gaussian Mixture Model. The 'mx assign' algorithm operates on a submatrix of marker genes, like standard algorithms such as CellAssign.
Mx assign performs assignment on a per matrix basis. mx assign also performs assignments on matrices normalized using ranks. This means the distance measurement via Euclidean distance in the GMM is replaced with the Spearman correlation.
Commons Cell Atlas (CCA), an infrastructure for the reproducible generation of a single cell atlas. CCA consists of a series of 'mx' and 'ec' commands that can modularly process count matrices. CCA can be constantly modified by the automated and continuous running of the tools.
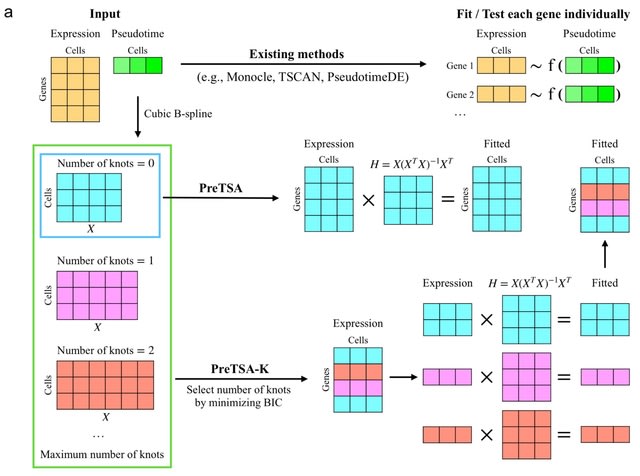
□ PreTSA: computationally efficient modeling of temporal and spatial gene expression patterns
>> https://www.biorxiv.org/content/10.1101/2024.03.20.585926v1
PreTSA (Pattern recognition in Temporal and Spatial Analyses) fits a regression model using B-splines, obtaining a smoothed curve that represents the relationship between gene expression values and pseudotime.
PreTSA performs all computations related to the design matrix once. PreTSA leverages efficient matrix operations in R to further enhance computational efficiency. By default, PreTSA employs the simplest B-spline basis without internal knots to achieve optimal computational speed.
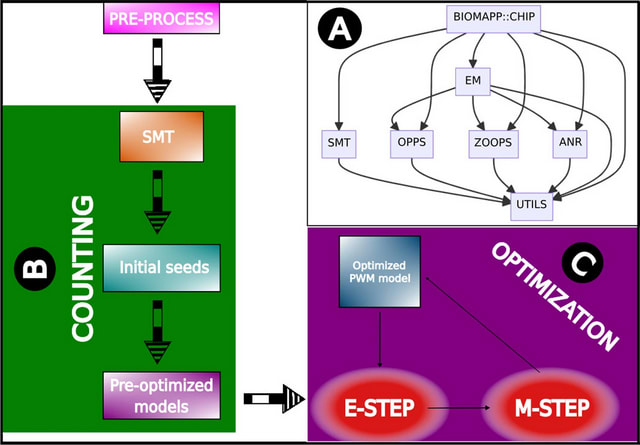
□ BIOMAPP::CHIP: large-scale motif analysis
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05752-3
BIOMAPP::CHIP (Biological Application for ChIP-seq data) adopts a two-step approach for motif discovery: counting and optimization. In the counting phase, the SMT (Sparse Motif Tree) is employed for efficient kmer counting, enabling rapid and precise analysis.
BIOMAPP::CHIP loads the pre-processed data, which contains the nucleotide sequences of interest. Next, if available, control sequences are loaded. Otherwise, they are generated by shuffling the main dataset using the MARKOV method or the EULER method.
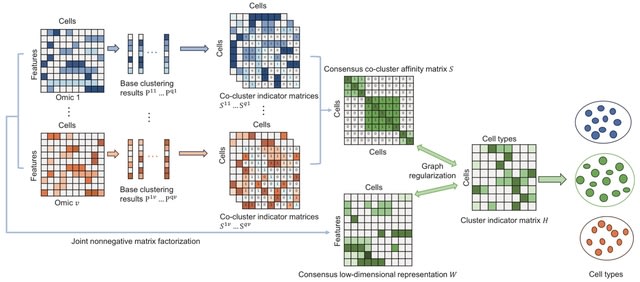
□ GRMEC-SC: Clustering single-cell multi-omics data via graph regularized multi-view ensemble learning
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae169/7636963
GRMEC-SC (Graph Regularized Multi-view Ensemble Clustering) can simultaneously learn the consensus low-dimensional representations and the consensus co-cluster affinity matrix of cells from multiple omics data and multiple base clustering results.
There are two trade-off hyper-parameters λ1 and λ2 in the GRMEC-SC model. λ1 controls the agreement between the low-dimensional representation W and the cluster indicator matrix H, while λ2 controls the effect of the consensus co-cluster affinity matrix S on the final clustering.

□ Accelerated dimensionality reduction of single -cell RNA sequencing data with fastglmpca
>> https://www.biorxiv.org/content/10.1101/2024.03.23.586420v1
Alternating Poisson Regression (APR), has strong convergence guarantees; the block-coordinatewise updates monotonically improve the log-likelihood, and under mild conditions converge to a (local) maximum of the likelihood.
In addition, by splitting the large optimization problem into smaller pieces (the Poisson GLMs), the computations are memory-efficient and are trivially parallelized to take advantage of multi-core processors.
Since APR reduces the problem of fitting a Poisson GLM-PCA model to the problem of fitting many (much smaller) Poisson GLMs, the speed of the APR algorithm depends critically on how efficiently one can fit the individual Poisson GLMs.

□ scCASE: accurate and interpretable enhancement for single-cell chromatin accessibility sequencing data
>> https://www.nature.com/articles/s41467-024-46045-w
scCASE takes a preprocessed scCAS count matrix as input, then generates an initial similarity matrix based on the matrix and performs non-negative matrix factorization to obtain an initial projection matrix and an initial cell embedding.
Random sampling matrix is generated through binomial distribution and Hadamard multiplied with similarity matrix in the computation is to avoid same cells exhibit almost the same accessible peaks which improperly reduces the cellular heterogeneity.
scCASE model uses similarity and matrix factorization to enhance scCAS data separately and iteratively optimizes the initialized matrix, aiming to minimize the difference between the reconstructed and enhanced matrices.
□ AutoXAI4Omics: an Automated Explainable AI tool for Omics and tabular data
>> https://www.biorxiv.org/content/10.1101/2024.03.25.586460v1
AutoXAI4Omics performs regression tasks from omics and tabular numerical data. AutoXAI4Omics encompasses an automated workflow that takes the user from data ingestion through data preprocessing and automated feature selection to generate a series of hyper-tuned optimal ML models.
AutoXAI4Omics facilitates interpretability of results, not only via feature importance inference, but using XAI to provide the user with a detailed global explanation of the most influential features contributing to each generated model.
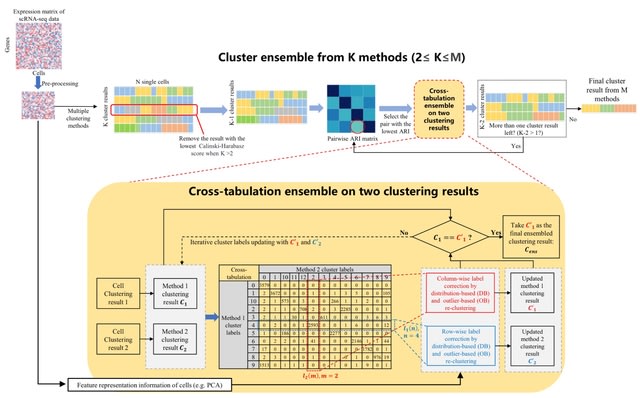
□ CTEC: a cross-tabulation ensemble clustering approach for single-cell RNA sequencing data analysis
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btae130/7637679
CTEC can integrate a pair of clustering results by taking advantage of two SOTA methods, the community detection-based Leiden method and the unsupervised Deep Embedding algorithm that clusters Single-Cell RNA-seq data by iteratively optimizing a clustering objective function.
A cross-tabulation of the clustering results from the two individual methods is generated. After that, one of two correction schemes is carried out under different circumstances: outlier-based clustering correction and distribution-based clustering correction.

□ Optimizing ODE-derived Synthetic Data for Transfer Learning in Dynamical Biological Systems
>> https://www.biorxiv.org/content/10.1101/2024.03.25.586390v1
A systematic approach for optimizing the ODE-based synthetic dataset characteristics for time series forecasting in a simulation-based TL setting. This framework allows selecting characteristics of ODE-derived datasets and their multivariate investigation.
This framework generates synthetic datasets with different characteristics: They consider the synthetic dataset size, the diversity of the ODE dynamics represented in the synthetic data, and the synthetic noise applied to the data.
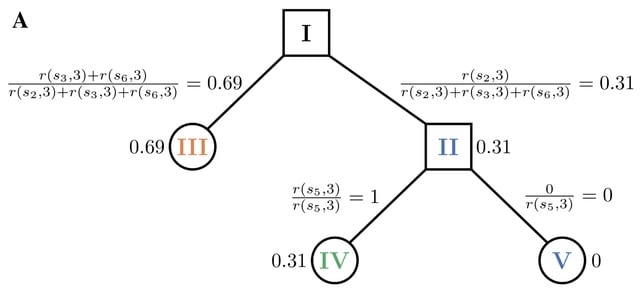
□ tangleGen : Inferring Ancestry with the Hierarchical Soft Clustering Approach
>> https://www.biorxiv.org/content/10.1101/2024.03.27.586940v1
tangleGen infers population structures in population genetics and exploit its capabilities. tangleGen is based on Tangles, a theoretical concept that originated from mathematical graph theory, where they were initially conceptualized to represent highly cohesive structures.
Tangles aggregates information about the data set's structure from many bipartitions that give local insight. Thereby, the information from many weaker, imperfect bipartitions is combined into an expressive clustering.
tangleGen uses bipartitions of genetic data to achieve a hierarchical soft clustering. tangleGen also adds a new level of explainability, as it extracts the relevant SNPs of the clustering.

□ Cellects, a software to quantify cell expansion and motion
>> https://www.biorxiv.org/content/10.1101/2024.03.26.586795v1
Cellects, a tool to quantify growth and motion in 2D. This software operates with image sequences containing specimens growing and moving on an immobile flat surface.
Cellects provides the region covered by the specimens at each point of time, as well as many geometrical descriptors that characterize it. Cellects works on images or videos containing multiple arenas, which are automatically detected and can have a variety of shapes.
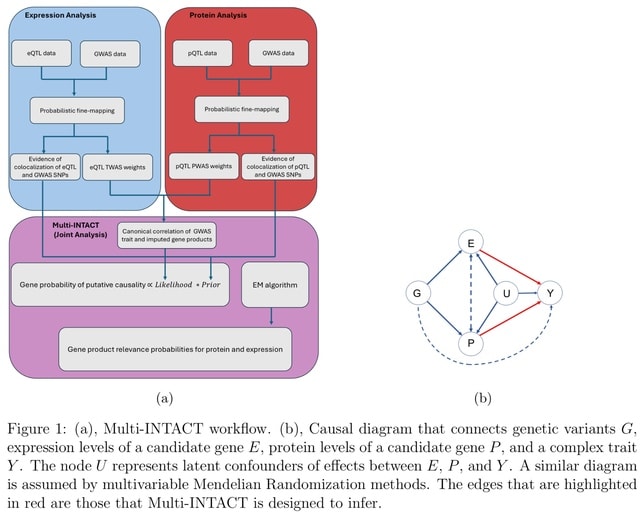
□ Multi-INTACT: Integrative analysis of the genome, transcriptome, and proteome identifies causal mechanisms of complex traits.
>> https://www.biorxiv.org/content/10.1101/2024.03.28.587202v1
Multi-INTACT leverages information from multiple molecular phenotypes to implicate putative causal genes (PCGs). Multi-INTACT gauges the causal significance of a target gene concerning a complex trait and identifies the pivotal gene products.
Multi-INTACT extends the canonical single-exposure (i.e., a single molecular phenotype) instrumental variables (IV) analysis/TWAS method to account for multiple endogenous variables, integrating colocalization evidence in the process.










※コメント投稿者のブログIDはブログ作成者のみに通知されます