(Created with Midjourney v5.2)

□ scNODE: Generative Model for Temporal Single Cell Transcriptomic Data Prediction
>> https://www.biorxiv.org/content/10.1101/2023.11.22.568346v1
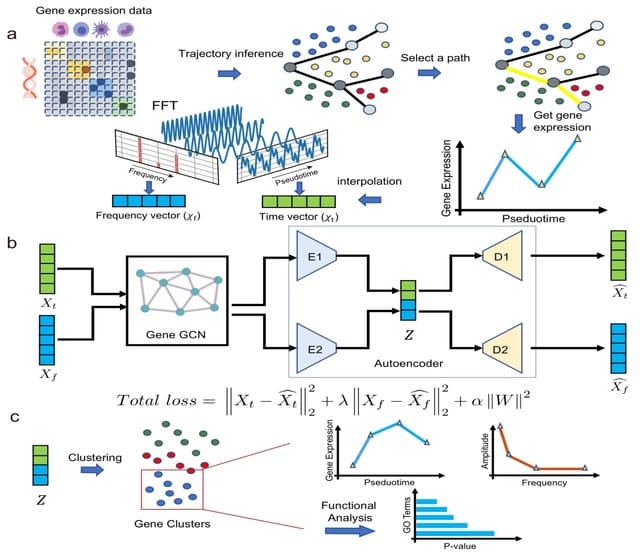
scNODE (single-cell neural ODE) is a generative model that simulates and predicts realistic in silico single-cell gene expressions at any timepoint. scNODE integrates the VAE and neural ODE to model cell developmental landscapes on the non-linear manifold.
scNODE constructs a most probable path between any two points through the Least Action Path (LAP) method. The optimal path is not simply the algebraically shortest path in the gene expression space but follows the cell differential landscape in latent space modeled by scNODE.

□ Bert-Path: Integration of Multiple Terminology Bases: A Multi-View Alignment Method Using The Hierarchical Structure
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad689/7424708
Bert-Path, a multi-view framework that considers the semantic, neighborhood, and hierarchical features. Bert-Path involves incorporating interactive scores of the hierarchical paths into the alignment process, which reduces errors caused by differing levels between terminologies.
Bert-Path calculates the hierarchical differences between different entities in order to filter out entities with similar hierarchical paths. It employs a k-dimensional RBF kernel function. The alignment scores are obtained through an MLP with a gate mechanism.

□ BIOFORMERS: A SCALABLE FRAMEWORK FOR EXPLORING BIOSTATES USING TRANSFORMERS
>> https://www.biorxiv.org/content/10.1101/2023.11.29.569320v1
BioFormers is inspired by scGPT and scBERT to operate on the biostate of sample and phenotypical information of a sample. The biostate is defined as a high-dimensional vector that includes various biological markers.
During the experiments, they also train the model on value-binned data that are not normalized in order to explore the impact of normalization and the variance in the "semantic" meaning of gene expression counts.
BioFormers may retrieve general biological knowledge in a zero-shot learning process. BioFormers allows for the inclusion of external tokens, which carry meta-information related to individual molecules.

□ GSPA: Mapping the gene space at single-cell resolution with gene signal pattern analysis
>> https://www.biorxiv.org/content/10.1101/2023.11.26.568492v1
GSPA (gene signal pattern analysis), a new method for embedding genes in single-cell datasets using a novel combination of diffusion wavelets and deep learning. GSPA builds a cell-cell graph and define any genes measured as signals on the cell-cell graph.
GSPA decomposes the gene signal using a large dictionary of diffusion wavelets of varying scales that are placed at different locations on the graph. The result is a representation of each gene in a single-cell dataset as a set of graph diffusion wavelet coefficients.

□ GFETM: Genome Foundation-based Embedded Topic Model for scATAC-seq Modeling
>> https://www.biorxiv.org/content/10.1101/2023.11.09.566403v1
GFTM, an interpretable and transferable deep neural network framework that integrates GFM and Embedded Topic Model (ETM) to perform scATAC-seq data analysis. In the zero-shot transfer setting, the GFETM model was first trained on a source scATAC-seq dataset.
GFETM is designed to jointly train ETM and GFM. The ETM comprises an encoder and a linear decoder that encompass topic embeddings, peak embeddings, and batch effect intercepts. In parallel, the GFM takes the DNA sequences of peaks as inputs and generates sequence embeddings.
Each scATAC-seq profile serves as an input to a variational autoencoder (VAE) as the normalized peak count. The encoder network produces the latent topic mixture for clustering cells.
The GFETM model takes the peak sequence as input and output peak embeddings. The linear decoder learns topic embedding to reconstruct the input. The encoder, decoder and genome fondation model are jointly optimized by maximizing ELBO.

□ Flowtigs: safety in flow decompositions for assembly graphs
>> https://www.biorxiv.org/content/10.1101/2023.11.17.567499v1
Flowtigs, a linear-time-verifiable complete characterisation of walks that are safe in flow decompositions, i.e. that are subwalks of any possible flow decomposition.
Flowtigs generalises over the previous one for DAGs, using a more involved proof of correctness that works around various issues introduced by cycles.
Providing an optimal O(mn)-time algorithm that identifies all maximal flowtigs and represents them inside a compact structure. Flowtigs use all information that is available through the structure of the assembly graph and the abundance values on the arcs.

□ Haplotype-aware Sequence-to-Graph Alignment
>> https://www.biorxiv.org/content/10.1101/2023.11.15.566493v1
The 'haplotype-aware' formulations for sequence-to-DAG alignment and sequence-to-DAG chaining problems. This formulations use the haplotype path information available in modern pangenome graphs. The formulations are inspired from the classic Li-Stephens haplotype copying model.
The Li-Stephens model is a probabilistic generative model which assumes that a sampled haplotype is an imperfect mosaic of known haplotypes. Similarly, this haplotype-aware sequence-to-DAG alignment formulation optimizes the number of edits and haplotype switches simultaneously.
An alignment path specifies a path in the DAG and the indices of the selected haplotypes along the path. Formulating haplotype-aware co-linear chaining problem. They solve it in O(|H|Nlog|H|N) time, assuming a one-time O|E||H|) indexing of the DAG.

□ MetageNN: a memory-efficient neural network taxonomic classifier robust to sequencing errors and missing genomes
>> https://www.biorxiv.org/content/10.1101/2023.12.01.569515v1
MetageNN is a neural network model that uses short k-mer profiles of sequences to reduce the impact of distribution shifts on error-prone long reads. By utilizing nanopore sequencing data, MetageNN exhibits improved sensitivity in situations where the reference database is incomplete.
MetageNN surpasses the alignment-based MetaMaps and MEGAN-LR, as well as the k-mer-based Kraken2 tools, with improvements of 100%, 36%, and 23% respectively at the read-level analysis.


□ JEM-mapper: An Efficient Parallel Sketch-based Algorithmic Workflow for Mapping Long Reads
>> https://www.biorxiv.org/content/10.1101/2023.11.28.569084v1
JEM-mapper, an efficient parallel algorithmic workflow that uses a new minimizer-based Jaccard estimator (or JEM) sketch to perform alignment-free mapping of long reads.
The JEM-mapper algorithm can be used to map long reads to either a set of partially assembled contigs (from a previous short read assembly), or to the set of long reads themselves.

□ Isosceles: Accurate long-read transcript discovery and quantification at single-cell resolution with Isosceles
>> https://www.biorxiv.org/content/10.1101/2023.11.30.566884v1
Isosceles (the Isoforms from single-cell, long-read expression suite); a computational toolkit for reference-guided de novo detection, accurate quantification, and downstream analysis of full-length isoforms at either single-cell, pseudo-bulk, or bulk resolution levels.
Isosceles achieves multi-resolution quantification by using the EM algorithm. Isosceles utilizes acyclic splice-graphs to represent gene structure. In the graph, nodes represent exons, edges denote introns, and paths through the graph correspond to whole transcripts.

□ Polygraph: A Software Framework for the Systematic Assessment of Synthetic Regulatory DNA Elements
>> https://www.biorxiv.org/content/10.1101/2023.11.27.568764v1
Polygraph provides a variety of features to streamline the synthesis and scrutiny of regulatory elements, incorporating features like a diversity index, motif and k-mer composition, similarity to endogenous regulatory sequences, and screening with predictive and foundational models.
Polygraph uses HyenaDNA to quantify the log likelihood of synthetic sequences to score their "humanness". A sequence diversity metric is defined as the average KNN distance between a sequence and its neighbors, to quantify how similar designed sequences are to each other.

□ TREVI: A Transcriptional Regulation-driven Variational Inference Model to Speculate Gene Expression Mechanism with Integration of Single-cell Multi-omics
>> https://www.biorxiv.org/content/10.1101/2023.11.22.568363v1
TREVIXMBD (Transcriptional REgulation-driven Variational Inference) devises a Bayesian framework to incorporate the well-established gene regulation structure. TREVIXMBD triggers the generation process for gene expression profile and infers the latent variables.
TREVIXMBD aims to optimize the estimation of TF activities and the TF-gene interactions by precisely modeling the generation of single-cell profiles under the synergistic control of TFs and other genetic elements.

□ HERO: Hybrid-hybrid correction of errors in long reads
>> https://www.biorxiv.org/content/10.1101/2023.11.10.566673v1
HERO (Hybrid Error coRrectiOn) is "hybrid-hybrid" insofar as it uses both NGS + TGS reads, so is hybrid in terms of using reads w/ complemenentary properties, and both DBG's + MA's/OG's on the other hand, so is hybrid w/ respect to the employment of complementary data structures.
The foundation of HERO is the idea that aligning the short NGS reads with the long TGS reads prior to correction yields corrupted alignments because of the abundantly occurring indel artifacts in the TGS reads.
HERO aligns NGS reads with (DBG based pre-corrected) TGS reads, and then uses the TGS read as a template for phasing the NGS reads that align with them, and subsequently discarding the NGS reads that do not agree with the TGS template read in terms of phase.
HERO pre-phases the long TGS reads prior to aligning them with the NGS reads. If pre-phased sufficiently well, TGS reads get aligned only with NGS reads that stem from the same phase, which avoids the time consuming filtering out of spurious NGS-TGS alignments.

□ NeuroVelo: interpretable learning of cellular dynamics from single-cell transcriptomic data
>> https://www.biorxiv.org/content/10.1101/2023.11.17.567500v1
NeuroVelo combines ideas from Neural Ordinary Differential Equations (ODE) and RNA velocity in a physics-informed neural network architecture. NeuroVelo uses a novel rank-based statistic to provide a robust way to identify genes associated w/ dynamical changes in cellular state.
NeuroVelo model has two autoencoders, one is a non-linear 1D encoder learning a pseudo-time coordinate associated with each cell, while the second is a linear projection to an effective phase space for the system.

□ The bulk deep generative decoder: N-of-one differential gene expression without control samples using a deep generative model
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03104-7
bulkDGD is based on the Deep Generative Decoder (DGD), a generative neural network that learns a probabilistic low-dimensional representation of the data. The model is trained on the Genotype-Tissue Expression (GTEx) database maps the latent space to the data space.
bulkDGD learns the most probable representation for each sample in the low-dimensional space. A fully connected feed-forward decoder neural network with two hidden layers maps the latent space to sample space, resulting in a negative binomial distribution for each gene.

□ GENTANGLE: integrated computational design of gene entanglements
>> https://www.biorxiv.org/content/10.1101/2023.11.09.565696v2
GENTANGLE (Gene Tuples ArraNGed in overLapping Elements) is a high performance containerized pipeline for the computational design of two overlapping genes translated in different reading frames of the genome that can be used to design gene entanglements.
The GENTANGLE pipeline includes newly developed software to visualize and select CAMEOX sequence proposals. Each candidate solution plots the negative pseudo loglikelihood (NPLL) scores predicting the fitness potential of each protein in the entangled gene.
Additional information for each solution includes sequence similarity between the synthetic sequence and wild type, and the relative starting position of the shorter gene embedded in the longer gene referred to as the Entanglement Relative Position (ERP).
The NPLL space is searched for a tentative number of non-overlapping ranges corresponding to a higher density of variants while maximizing the pairwise distance of the range's centers of mass.
The NPLL scores are initially grouped into discrete bins with similarly scored solutions with the goal of making a balanced selection of proposed solutions across the span of predicted fitness values.

□ Comparing methods for constructing and representing human pangenome graphs
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03098-2
A comprehensive view of whole-genome human pangenomics through the lens of five methods that each implement a different graph data structure: Bifrost, Minimizer-space de Bruijn graphs (mdbg), Minigraph, Minigraph-Cactus, and PanGenome Graph Builder (pggb).
pggb is a directed acyclic variation graph construction pipeline. It calls three different tools: pairwise base-level alignment of haplotypes using wfmash, graph construction from the alignments with seqwish, graph sorting and normalization with smoothxg and GFAffix.
pggb facilitates downstream analyses using the companion tool odgi. Minigraph generates a pangenome graph based on a reference sequence taken as a backbone. It shines in the representation of complex structural variations, but does not incl. small or inter-chromosomal variations.
The pipeline Minigraph-Cactus, which uses the Cactus base aligner, can be used to add small-level variations on top of the Minigraph graph and to keep a lossless representation of the input sequences.
Bifrost illustrates that classical de Bruijn graphs are scalable, stable, dynamic, and store all variations. mdbg is the fastest construction method which generates an approximate representation of differences between haplotypes.

□ IDESS: a toolbox for identification and automated design of stochastic gene circuits
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad682/7439590
DESS (Identification and automated DEsign of Stochastic gene circuitS), is capable of simulating stochastic biocircuits very efficiently using GPU acceleration for simulation and global optimization.
IDESS includes CPU and GPU parallel implementations of the Stochastic Simulation Algorithm (SSA) and the semi-Lagrangian Simulation method in SELANSI. This semi-Lagrangian numerical method simulates a Partial Integro-Differential Equation model describing the biocircuit dynamics.
IDESS utilizes Global Optimization solvers capable of optimizing over high dimensional search spaces of continuous real and discrete integer variables, including Mixed Integer Nonlinear Programming solvers to optimize simultaneously across parameter and topology search spaces.

□ Sylph: Metagenome profiling and containment estimation through abundance-corrected k-mer sketching
>> https://www.biorxiv.org/content/10.1101/2023.11.20.567879v1
sylph, a metagenome profiler that estimates metagenome-genome average nucleotide identity (ANI) through zero-inflated Poisson k-mer statistics, enabling ANI-based taxa detection.
Sylph transforms a database of reference genomes and a metagenome into subsampled k-mers using FracMinHash, sampling approximately one out of c k-mers (c = 200 by default). Sylph then analyzes the containment of the genomes' k-mers in the metagenome.

□ scLongTree: an accurate computational tool to infer the longitudinal tree for scDNAseq data
>> https://www.biorxiv.org/content/10.1101/2023.11.11.566680v1
scLongTree, a computational tool to infer the longitudinal subclonal tree based on the longitudinal scDNA-seq data from multiple time points. Different from LACE, scLong Tree does not hold a ISA and thus allows parallel and back mutations.
scLongTree reconstructs unobserved subclones that are not represented by any cells sequenced. By adopting a myriad of statistical methods as well as corroborating the cells all across distinct time points, scLongTree is able to identify spurious subclones and eliminate them.
ScLongTree’s tree inference algorithm is sophisticated in the sense that it can infer up to two levels of unobserved nodes in between two consecutive time points, and it searches for a tree with the least number of back mutations and parallel mutations.
scLongTree infers a longitudinal tree that connects the subclones among different time points, and places the mutations on the edges. If necessary, scLongTree adds the unobserved nodes in between two consecutive time points.

□ Sketching methods with small window guarantee using minimum decycling sets
>> https://arxiv.org/abs/2311.03592
A Minimum Decycling Set (MDS) is a set of k-mers that is unavoidable and of minimum size. MDSs provide a logical starting point for the study of decycling sets. The MDSs are by definition as small as possible, therefore reducing as much as possible the cost of querying a set.
An optimization procedure is designed to find MDSs with short remaining path lengths. This optimization procedure gives further insight on the range of possible window guarantee for sketching methods and on the of the well-known Mykkeltveit set.

□ PathExpSurv: pathway expansion for explainable survival analysis and disease gene discovery
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05535-2
PathExpSurv, a novel survival analysis method by exploiting and expanding the existing pathways. They added the genes beyond the databases into the NN pre-trained using the existing pathways, and continued to train a regularized survival analysis model, with a L1 penalty.
PathExpSurv can gain an insight into the black-box model of neural network for survival analysis. PathExpSurv a novel optimization scheme consisting 2 phases: pre-training / training phase, in order to improve the performance of neural network by expanding the prior pathways.

□ SPREd: A simulation-supervised neural network tool for gene regulatory network reconstruction
>> https://www.biorxiv.org/content/10.1101/2023.11.09.566399v1
SPREd (Supervised Predictor of Regulatory Edges), utilizes a neural network to relate an expression matrix to the corresponding GRN. GRNs are constructed based on the feature importance of TFs (features) in the model trained for a target gene.
In SPREd, an ML model is trained to directly predict TFs regulating a target gene, based on expression matrix of all TFs and the target gene. The ML model is trained on simulated expression matrix-GRN pairs and can then be used to predict the GRN for any expression matrix.

□ L1-regularized DNN estimator: Statistical learning by sparse deep neural networks
>> https://arxiv.org/abs/2311.08845
A deep neural network estimator based on empirical risk minimization with L1-regularization. It derives a general bound for its excess risk in regression, and prove that it is adaptively nearly-minimax simultaneously across the entire range of various function classes.
The minimax convergence rates over various function classes suffer from a well-known curse of dimensionality phenomenon. To reduce the large number of parameters in a fully-connected DNN one can consider specific types of sparse architectures.
There are several possible ways to define DNN sparsity: connection sparsity (small number of active connections between nodes), one can consider other notions of sparsity, e.g. node sparsity (small number of active nodes) and layer sparsity.

□ Speeding up iterative applications of the BUILD supertree algorithm
>> https://www.biorxiv.org/content/10.1101/2023.11.10.566627v1
This version of the BUILD algorithm constructs the connected components of the cluster graph without explicitly constructing the cluster graph. That is, this algorithm does not directly represent the edges of the cluster graph in memory.
The fully incrementalized algorithm BUILDINC adds the ability to track changes that are made to the solution object, and then roll them back if the algorithm ultimately returns FALSE.

□ Recomb-Mix: Fast and accurate local ancestry inference
>> https://www.biorxiv.org/content/10.1101/2023.11.17.567650v1
Recomb-MIX, a novel local ancestry inference (LAI) method that integrates the elements of existing methods and introduces a new graph collapsing to simplify counting paths with the same ancestry label readout.
Recomb-Mix enables the collapsing of the reference panel to a compact graph. Generating a compact graph greatly reduces the size of reference populations and retains the ancestry information as most non-ancestry informative markers are collapsed in the compact graph.
Different path change penalties were used when switching haplotype templates: the path change penalty within a reference population is set to zero, and the path change penalty between the reference populations is parameterized by recombination rates from a genetic map.

□ ROCCO: A Robust Method for Detection of Open Chromatin via Convex Optimization
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad725/7455257
ROCCO determines consensus open chromatin regions across multiple samples simultaneously. ROCCO employs robust summary statistics and solves a constrained optimization problem formulated to account for both enrichment and spatial dependence of open chromatin signal data.
ROCCO accounts for features common to the edges of accessible chromatin regions, which are often hard to determine based on independently determined sample peaks that can vary widely in their genomic locations.

□ TsImpute: An accurate two-step imputation method for single-cell RNA-seq data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad731/7457483
TsImpute adpots zero-inflated negative binomial distribution to discriminate dropouts from true zeros and performs initial imputation by calculating the expected expression level.
TsImpute calculates the Euclidean distance matrix based on the imputed expression matrix and adopts inverse distance weighed imputation to conduct the final imputation.

□ CIA: a Cluster Independent Annotation method to investigate cell identities in scRNA-seq data
>> https://www.biorxiv.org/content/10.1101/2023.11.30.569382v1
Given a set of gene signatures in Gene Matrix Transposed (GMT) file format and a gene expression matrix in an AnnData object, CIA builds a score matrix with signature scores for each entry in the gene signature file and every cell in the expression matrix.

□ Minimizing Reference Bias with an Impute-First Approach
>> https://www.biorxiv.org/content/10.1101/2023.11.30.568362v1
A novel impute-first alignment framework that combines elements of genotype imputation and pangenome alignment. It begins by genotyping the individual from a subsample of the input reads.
The workflow.indexes the personalized reference and applies a read aligner, which could be a linear or graph aligner, to align the full read set to the personalized reference.
The workflow is modular; different tools can be substituted for the initial genotyping step (e.g. Bowtie2+bcftools instead of Rowbowt), the imputation step (e.g. Beagle instead of Glimpse) and the final read alignment step (e.g. Bowtie2 or BWA-MEM instead of VG Giraffe).