□ Stabilization of extensive fine-scale diversity by spatio-temporal chaos
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/15/736215.full.pdf
Enormous diversity of species is one of the remarkable features of life on Earth, the antisymmetric correlations in the Lotka-Volterra interaction matrix, together with simple spatial structure, are sufficient to stabilize extensive diversity of an assembled community.
the spatio-temporally chaotic “phase” should exist in a wide range of models, and that even in rapidly mixed systems, longer lived spores could similarly stabilize a diverse chaotic phase.

□ SigMa: Hidden Markov models lead to higher resolution maps of mutation signature activity
>> https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-019-0659-1
The SigMa model has two components: sky mutations using a multinomial mixture model (MMM). The MMM is characterized by a vector g of K mutation signature marginal probabilities and the same emission matrix E.
To model cloud mutations, using a dynamic Bayesian network (DBN) that is a simple extension of an HMM in that it allows subsequences generated by the HMM to be interspersed with mutations generated by the MMM.
SigMa finds the most likely sequence of signatures that explains the observed mutations in sky and clouds.
□ Signac: an extension of Seurat for the analysis, interpretation, and exploration of single-cell chromatin datasets
>> https://satijalab.org/signac/index.html
Signac is currently focused on the analysis of single-cell ATAC-seq data, but new features will be added as experimental methods for measuring other chromatin-based modalities at single-cell resolution become more widespread.
Signac calculates single-cell QC metrics, multiple single-cell ATAC-seq datasets, and also visualizing ‘pseudo-bulk’ coverage tracks.

□ Cactus: Progressive alignment - a multiple-genome aligner for the thousand-genome era
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/09/730531.full.pdf
Cactus, a reference-free multiple genome alignment program, has been shown to be highly accurate, but the existing implementation scales poorly with increasing numbers of genomes, and struggles in regions of highly duplicated sequence.
Cactus is capable of scaling to hundreds of genomes and beyond by describing results from an alignment of over 600 amniote genomes, which is to this knowledge the largest multiple vertebrate genome alignment yet created.

□ Inferring reaction network structure from single-cell, multiplex data, using toric systems theory
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/09/731018.full.pdf
This application of toric theory enables a data-driven mapping of covariance relationships in single cell measurements into stoichiometric information, one in which each cell subpopulation has its associated ESS interpreted in terms of CRN theory.
For limit cycles, the expected geometry is not necessarily algebraic, although one could hope that the limit cycle is contained in an almost-toric manifold, so that our approach is still informative.

□ GePhEx: Genome-Phenome Explorer: A tool for the visualization and interpretation of phenotypic relationships supported by genetic evidence
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz622/5545092
GePhEx complements the list of SNPs adding variants in LD and the corresponding associated traits. Finally, GePhEx infers the phenotypic relationships supported by genetic data considering the list of SNPs and the associated traits.
GePhEx does retrieve well-known relationships as well as novel ones, and that, thus, it might help shedding light on the pathophysiological mechanisms underlying complex diseases.

□ MEPSAnd: Minimum Energy Path Surface Analysis over n-dimensional surfaces
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz649/5550624
MEPSAnd, an open source GUI-based program that natively calculates minimum energy paths across energy surfaces of any number of dimensions.
Among other features, MEPSAnd can compute the path through lowest barriers and automatically provide a set of alternative paths.
□ BBKNN: Fast Batch Alignment of Single Cell Transcriptomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz625/5545955
BBKNN (batch balanced k nearest neighbours) is a fast and intuitive batch effect removal tool that can be directly used in the scanpy workflow.
BBKNN outputs can be immediately used for dimensionality reduction, clustering and pseudotime trajectory inference.

□ BayICE: A hierarchical Bayesian deconvolution model with stochastic search variable selection
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/12/732743.full.pdf
a comprehensive Markov chain Monte Carlo procedure through Gibbs sampling to estimate cell proportions, gene expression profiles, and signature genes.
BayICE integrates gene expression deconvolution and gene selection in the same model, and incorporates SSVS, a Bayesian variable selection approach, to implement internal gene selection.

□ GAMBIT: Integrating Comprehensive Functional Annotations to Boost Power and Accuracy in Gene-Based Association Analysis https://www.biorxiv.org/content/biorxiv/early/2019/08/12/732404.full.pdf
a statistical framework and computational tool to integrate heterogeneous annotations with GWAS summary statistics for gene-based analysis, applied with comprehensive coding and tissue-specific regulatory annotations.
GAMBIT (Gene-based Analysis with oMniBus, Integrative Tests) is an open-source tool for calculating and combining annotation-stratified gene-based tests using GWAS summary statistics: single-variant association z-scores.

□ Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome
>> https://www.nature.com/articles/s41587-019-0217-9
the optimization of circular consensus sequencing (CCS) to improve the accuracy of single-molecule real-time (SMRT) sequencing (PacBio) and generate highly accurate (99.8%) long high-fidelity (HiFi) reads with an average length of 13.5 kilobases (kb).
De novo genome assembly using CCS reads alone produced a contiguous and accurate genome with a contig N50 of >15 megabases (Mb) and concordance of 99.997%, substantially outperforming assembly with less-accurate long reads.
□ pWGBSSimla: a profile-based whole-genome bisulfite sequencing data simulator incorporating methylation QTLs, allele-specific methylations and differentially methylated regions
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz635/5545541
pWGBSSimla is a profile-based whole-genome bisulphite sequencing data simulator, which can simulate WGBS, reduced representation bisulfite sequencing (RRBS), and oxidative bisulfite sequencing (oxBS-seq) data while modeling meQTLs, ASM, and differentially methylated regions.
□ Analyzing whole genome bisulfite sequencing data from highly divergent genotypes
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz674/5545001
a smoothing-based method that allows strain-unique CpGs to contribute to identification of differentially methylated regions (DMRs), and show that doing so increases power.
Map reads to personalized genomes and quantify methylation. Using whole-genome alignment tools, such as modmap or liftOver, place CpGs from each sample into a common coordinate space.
□ The bio.tools registry of software tools and data resources for the life sciences
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1772-6
enhancing the management of user profiles and crediting of contributions, e.g. using ELIXIR AAI federated user identity management, which incorporates researcher identities such as ORCID.
crosslink with portals such as ELIXIR TeSS (training resources) and FAIRSharing (data standards), in order to make navigation of the broader bioinformatics resource landscape more coherent.
□ Comparison of RNA Isolation Methods on RNA-Seq: Implications for Differential Expression and Meta-Analyses
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/12/728014.full.pdf
Within a self-contained experimental batch (e.g. control versus treatment), the method of RNA isolation had little effect on the ability to identify differentially expressed transcripts.
For meta-analyses however, researchers make every attempt to only compare experiments where the RNA isolation methods are similar.
□ openTSNE: a modular Python library for t-SNE dimensionality reduction and embedding
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/13/731877.full.pdf
openTSNE is still orders of magnitude faster than other Python im- plementations, including those from scikit-learn and MulticoreTSNE
define the affinity model based on two Gaussian kernels with varying perplexity, use a PCA- based initialization, and run the typical two-stage t-SNE optimization.
And reusing the embedding to map new data into existing embedding space.
embedding.affinities =
affinity.PerplexityBasedNN(
adata[:, genes].X, perplexity=30,
metric="cosine")
new_embedding = embedding.transform(data[:,
genes].X)
□ Transcriptome computational workbench (TCW): analysis of single and comparative transcriptomes
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/13/733311.full.pdf
The input to singleTCW is sequence and optional count files; the computations are sequence similarity annotation, gene ontology assignment, open reading frame (ORF) finding using hit information and 5th-order Markov models, and differential expression (DE).
TCW provides support for searching with the super-fast DIAMOND program against UniProt taxonomic databases, though the user can request BLAST and provide other databases to search against.
□ BERMUDA: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1764-6
BERMUDA (Batch Effect ReMoval Using Deep Autoencoders), a novel transfer-learning-based method for batch effect correction in scRNA-seq data.
BERMUDA effectively combines different batches of scRNA-seq data with vastly different cell population compositions and amplifies biological signals by transferring information among batches.
While BERMUDA was originally designed with a focus on scRNA-seq data with distinct cell populations, it can also accommodate such data by adjusting the resolution in the graph-based clustering algorithm and the trade-off between reconstruction loss and transfer loss to align clusters at a more granular level.

□ KSSD: Sequences Dimensionality-Reduction by K-mer Substring Space Sampling Enables Effective Resemblance- and Containment-Analysis for Large-Scale omics-data
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/14/729665.full.pdf
a new sequence sketching technique named k-mer substring space decomposition (kssd), which sketches sequences via k-mer substring space sampling instead of local-sensitive hashing.
illuminating the Jaccard- and the containment-coefficients estimated by kssd are essentially the sample proportions which are asymptotically gaussian distributed.
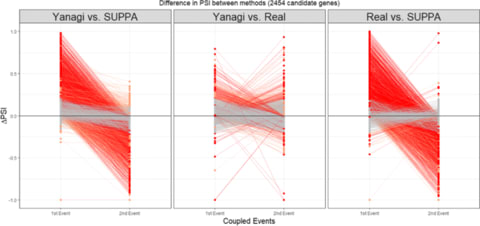
□ Yanagi: Fast and interpretable segment-based alternative splicing and gene expression analysis https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2947-6
Yanagi, an efficient algorithm to generate maximal disjoint segments given a transcriptome reference library on which ultra-fast pseudo-alignment can be used to produce per-sample segment counts.
□ LSX: automated reduction of gene-specific lineage evolutionary rate heterogeneity for multi-gene phylogeny inference
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3020-1
LSX includes a reprogrammed version of the original LS3 algorithm and has added features to make better lineage rate calculations.
the two modalities of the sequence subsampling algorithm included, LS3 and LS4, allow the user to optimize the amount of non-phylogenetic signal removed while keeping a maximum of phylogenetic signal.

□ DynOVis: a web tool to study dynamic perturbations for capturing dose-over-time effects in biological networks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2995-y
DynOVis allows studying both dynamic node expression changes and edge interaction changes simultaneously, whereas the current Cytoscape tools focus more on one topic.
With DynOVis offering the implementation of dynamic network visualization, by providing the users with functionalities to highlight node expression changes and dynamic edges.

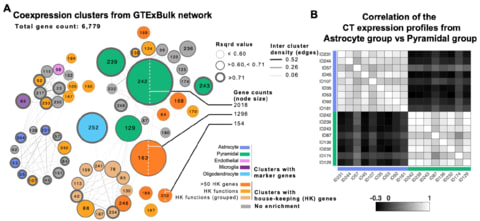
□ Untangling the effects of cellular composition on coexpression analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/15/735951.full.pdf
Beyond the implications for the goal of inferring regulation, the results have important implications for any use of expression data-based gene clustering or module identification in which the patterns are driven by cellular composition effects.
representation of the data as a network is potentially misleading, because it is tempting to interpret a network as representing physical relationships.
In particular, the idea that “hubs” in coexpression models are especially interesting is highly questionable if that pattern is simply a reflection of the cellular distribution of those transcripts.
□ Identifying and removing haplotypic duplication in primary genome assemblies
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/14/729962.full.pdf
a novel tool “purge_dups” that uses sequence similarity and read depth to automatically identify and remove both haplotigs and heterozygous overlaps.
after scaffolding w/ 10X Genomics linked reads using Scaff10x, purge_dups assembly generated 208 scaffolds w/ N50 23.82 Mb, and gap filling within the scaffolds during polishing w/ Arrow closed a substantial number of gaps, increasing contig N50 from 2.63 Mb initially to 14.50 Mb.
The scaffold and contig improvements were more modest when purge_haplotigs was used: 221 scaffolds with N50 8.17 Mb, and final contig N50 3.48 Mb. This indicates that divergent heterozygous overlaps can be a significant barrier to scaffolding, and that it is important to remove them as well as removing contained haplotigs.
□ Mercator: An R Package for Visualization of Distance Matrices
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/15/733261.full.pdf
Mercator implements several distance metrics between binary vectors, including Jaccard, Sokal-Michener, Hamming, Russell-Rao, Pearson, and Goodman-Kruskal.
Mercator provides access to four visualization methods, including hierarchical clustering, multidimensional scaling (MDS), t-distributed Stochastic Neighbor Embedding (t-SNE), and iGraph.

□ rawMSA: End-to-end Deep Learning using raw Multiple Sequence Alignments
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0220182
The core idea behind rawMSA is borrowed from the field of natural language processing to map amino acid sequences into an adaptively learned continuous space.
which they use to convert each residue character in the MSA into a floating-point vector of variable size.
This way of representing residues is adaptively learned by the network based on context, i.e. the structural property that trying to predict. And designing several deep neural networks based on this concept to predict SS, RSA, and Residue-Residue Contact Maps (CMAP).

□ Interpretable factor models of single-cell RNA-seq via variational autoencoders
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/16/737601.full.pdf
To investigate the potential for interpretability in the VAE framework, implementeing a linearly decoded variational autoencoder (LDVAE) in scVI.
interpretable non-Gaussian factor models can be linked to variational autoencoders to enable interpretable analysis of data at massive scale.
□ Multiple-kernel learning for genomic data mining and prediction
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2992-1
DALMKL optimizes the dual augmented Lagrangian of a proximal formulation of the MKL problem.
DALMKL formulation presents a unique set of problems such as the conjugate of a loss function must have no non-differentiable points in the interior of its domain and cannot have a finite gradient at the boundary of its domain.

□ FQStat: a parallel architecture for very high-speed assessment of sequencing quality metrics
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3015-y
FQStat is a stand-alone, platform-independent software tool that assesses the quality of FASTQ files using parallel programming.
FQStat uses a parallel programming architecture to automatic configuration of system parameters (e.g., core assignment and file segmentation) for optimum performance.

□ Query Combinators: Domain Specific Query Languages for Medical Research:
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/16/737619.full.pdf
The Observational Health Data Sciences and Informatics (OHDSI) cohort definition system is comprised of: a visual interface (Atlas), a JSON serialization format, which converts this JSON to SQL that is suitable to a particular database compliant with OHDSI’s Common Data Model.
Query Combinators can address this unmet need by enabling the cost-effective construction of domain specific query languages (DSQLs) specific to a research team.
□ Runnie: Run-Length Encoded Basecaller
>> https://github.com/nanoporetech/flappie/blob/master/RUNNIE.md
Runnie is an experimental basecaller that works in 'run-length encoded' space. Rather than calling a sequence of bases, where one call is one base, runs of bases (homopolymers) are called and so one call may represent many bases of the same type.
Run-length encoding separates a sequence into two parts: a run-length compressed sequencing containing only the identities of each run of bases, and the corresponding length of each run.
□ Telomere-to-telomere assembly of a complete human X chromosome
>> https://www.biorxiv.org/content/10.1101/735928v1
a de novo human genome assembly that surpasses the continuity of GRCh38, along with the first gapless, telomere-to-telomere assembly of a human chromosome.
This complete chromosome X, combined with the ultra-long nanopore data, also allowed us to map methylation patterns across complex tandem repeats and satellite arrays for the first time.
□ deSAMBA: fast and accurate classification of metagenomics long reads with sparse approximate matches
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/18/736777.full.pdf
deSAMBA, a tailored long read classification approach that uses a novel sparse approximate match block (SAMB)- based pseudo alignment algorithm.
It uses Unitig-BWT data structure to index the unitigs of the de Bruijn graph of the reference sequences, and finds similar blocks between reads and reference through the index.

□ SVFX: a machine-learning framework to quantify the pathogenicity of structural variants
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/19/739474.full.pdf
an agnostic machine-learning-based workflow, called SVFX, to assign a “pathogenicity score” to SVs in various diseases.
The model’s hyper-parameters (maximum depth of each tree in the forest, number of trees in the forest, and minimum number of leaves required to split an internal node) were tuned to maximize the Area Under the Receiver-Operator Curve and the Area Under the Precision Recall Curve.

□ Detecting selection from linked sites using an F-model
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/19/737916.full.pdf
an extension of F-model to linked loci by means of a hidden Markov model (HMM) that characterizes the effect of selection on linked markers through correlations in the locus specific component along the genome.
An obvious draw-back of modeling the locus-specific selection coefficients as a discrete Markov Chain is that for most candidate regions detected, multiple loci showed a strong signal of selection, making it difficult to identify the causal variant.
□ Multi-Scale Structural Analysis of Proteins by Deep Semantic Segmentation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz650/5551337
a Convolutional Neural Network that assigns each residue in a multi-domain protein to one of 38 architecture classes designated by the CATH database.
Semantic Segmentor is the trained classifier network, parser network, and entropy calculation.
□ SCATS: Detecting differential alternative splicing events in scRNA-seq with or without UMIs
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/19/738997.full.pdf
SCATS (Single-Cell Analysis of Transcript Splicing) for differential alternative splicing (DAS) analysis for scRNA-seq data with or without unique molecular identifiers (UMIs).
By modeling technical noise and grouping exons that originate from the same isoform(s), SCATS achieves high sensitivity to detect DAS events compared to Census, DEXSeq and MISO, and these events were confirmed by qRT-PCR experiment.
□ Swish: Nonparametric expression analysis using inferential replicate counts
>> https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz622/5542870
‘SAMseq With Inferential Samples Helps’, or Swish, that propagates quantification uncertainty from Gibbs posterior samples generated by the Salmon method for transcript quantification.
SWISH is a nonparametric model for differential expression analysis using inferential replicate counts, extending the existing SAMseq method to account for inferential uncertainty.

□ A model of pulldown alignments from SssI-treated DNA improves DNA methylation prediction
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3011-2
Against RRBS-determined methylation levels calculated genome-wide, BayMeth informed by the SssI pulldown model showed improvements as an indicator of methylated/unmethylated state, over BayMeth informed by observed SssI pulldown.
BayMeth with SssI data performed best among the three configurations, but BayMeth with our modeled SssI data always did better than BayMeth run without any SssI estimate.
□ Coverage profile correction of shallow-depth circulating cell free DNA sequencing via multi-distance learning
>> https://www.biorxiv.org/content/biorxiv/early/2019/08/20/737148.full.pdf
an empirically-driven coverage correction strategy that leverages prior annotation information in a multi-distance learning context to improve within-sample coverage profile correction.
a k-nearest neighbors (kNN) type of approach to leverage empirical bin-to-bin similarities and further integrate prior knowledge captured in genomic annotation sources via a multi-distance learning framework.
□ W.A.T.E.R.S.: a Workflow for the Alignment, Taxonomy, and Ecology of Ribosomal Sequences
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-317
WATERS employs Kepler system. Kepler system has a built-in database that allows calculations to be cached and stored internally rather than recalculated anew every time.